m6A修饰简介
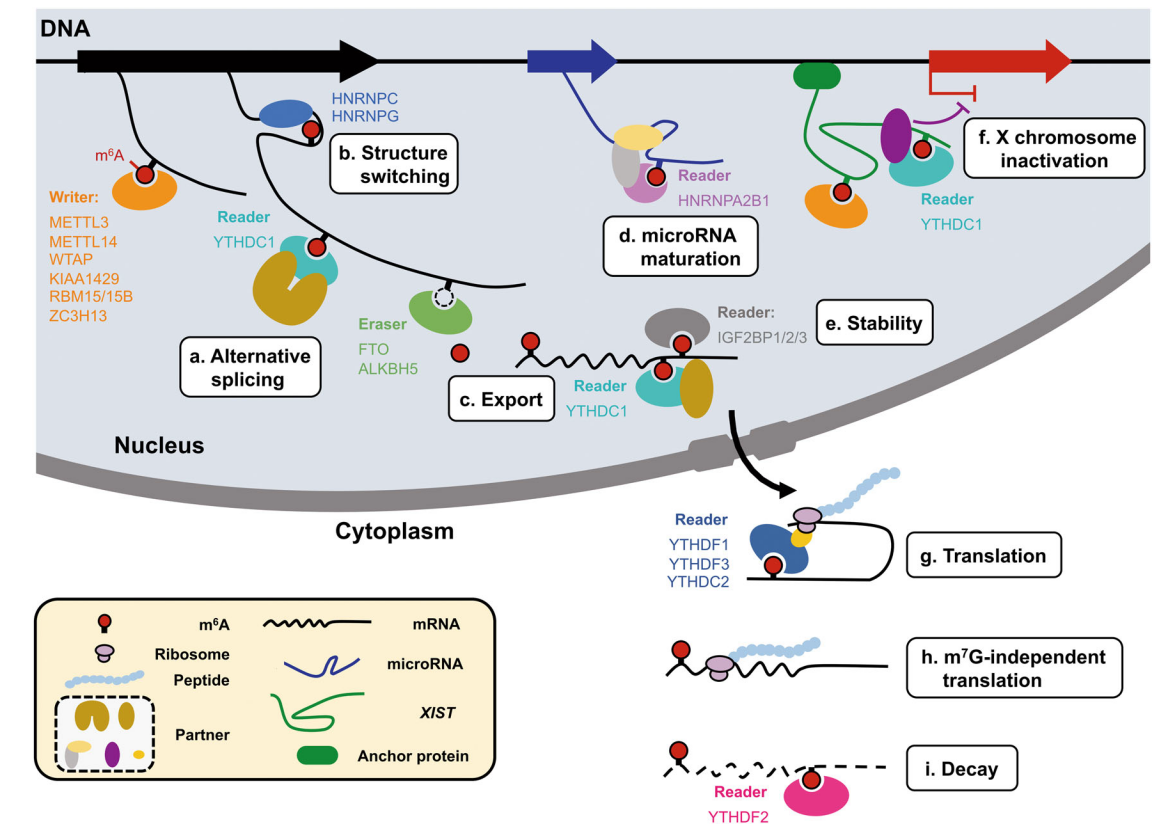
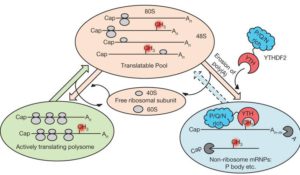
与表观遗传的DNA和组蛋白修饰一样,RNA也受到各种修饰,其中mRNA和lncRNA最主要的修饰是腺苷酸N6位的甲基化修饰(m6A)。m6A的修饰在真核生物中非常保守,从酵母、植物到高等动物中都广泛存在。在哺乳动物中,N6位被甲基化修饰的腺苷酸占细胞内所有mRNA腺苷酸的0.1-0.4%左右。RNA的m6A修饰是表观遗传学研究领域热点之一。在细胞内,甲基转移酶复合体(writers:METTL3及METTL14等)和去甲基化酶(erasers: FTO及ALKBH5)维持RNA的m6A水平的动态变化,而m6A识别蛋白(readers: YTHDFs等)特异性识别m6A位点,介导m6A的各种生物学功能。目前,大量研究已经证实m6A修饰具有重要功能:在分子功能层面,m6A修饰影响RNA的转录、加工、运输、翻译、降解等一系列过程;在生物学功能层面,m6A修饰参与调控生长发育、细胞分化、疾病发生发展等生物学过程(Yang et al. 2018)。因此,研究RNA的m6A修饰的动态变化,将在表观转录水水平,解析一些基本生物学问题,揭示疾病的发生发展机制,为寻找m6A调控的新药物靶点提供依据。
meRIP-seq简介及其技术原理
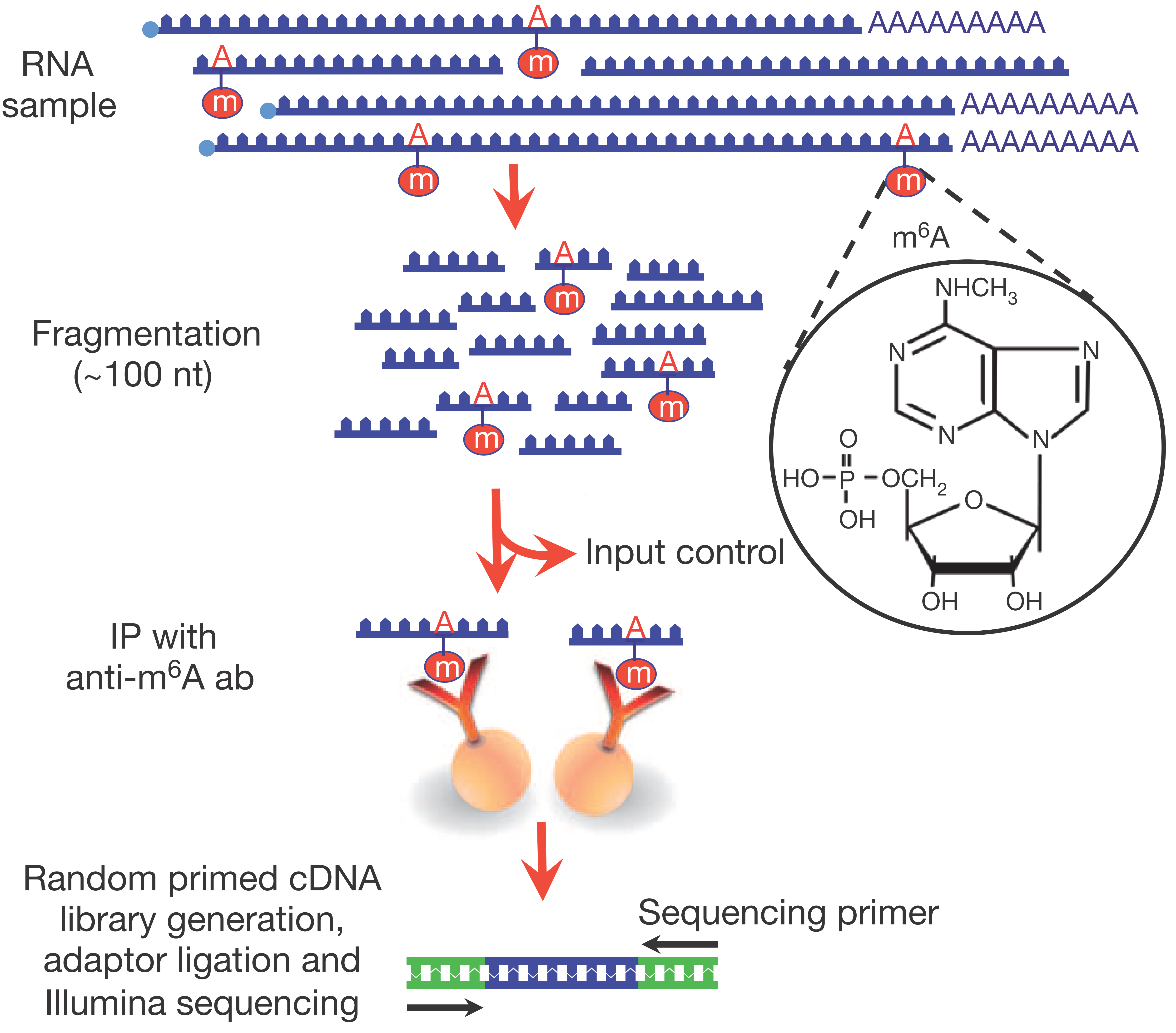
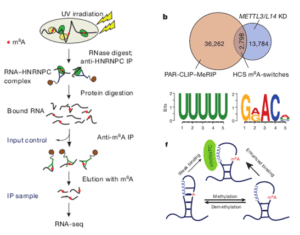
M6A修饰在人转录组RNA中广泛存在,目前研究表明,超过7000个mRNA和300个lncRNA都含有这种修饰;这种修饰富集分布在mRNA的终止密码子附近和3’UTR区域,暗示m6A修饰具有重要的调控功能。很多m6A修饰位点在人和小鼠中保守存在,且某些m6A修饰在不同发育时期,其修饰水平也在不断变化,同样暗示其重要的调控功能。目前对m6A的全基因组研究方法为meRIP-seq的方法,其原理为通过特异识别m6A修饰的抗体,对细胞内具有m6A修饰的RNA片段进行免疫共沉淀。通过对沉淀下来的RNA片段进行高通量测序,进而结合生物信息学分析,即可全基因组范围内对m6A修饰的状况进行系统研究。
产品优势
1、针对不同研究领域提供个性化方案;
2、分析流程全备,数据解读详细,后期数据挖掘可提供个性化分析;
3、结合峰分析策略多种:含Ablife和Piranha两种方法;
4、研究思路丰富:m6A修饰图谱及修饰特征分析,不同实验分组间差异m6A修饰位点和基因鉴定;与RNA-seq、CLIP-seq数据等联合分析,揭示m6A修饰的功能。
meRIP-seq适应样本类型及样品要求
1.样本类型:细胞、组织或RNA;
2.样本要求:RNA,大于10μg,浓度大于200ng/μl,质检合格;细胞干体积大于1000μl;组织样本大于或等于1000mg;
3.样品运输:样品置于1.5mL离心管中,封口膜封好,干冰运输。
4.样品保存:细胞样品或新鲜组织块可用TRIZOL或RNA保护剂处理,液氮冻存后-80℃保存;RNA样品可溶于乙醇或RNA-free的超纯水中,-80℃保存,避免反复冻融。
meRIP-seq的应该场景及分析思路
1. 应该场景:疾病、发育等生物学各方向;
2. 研究思路:
√ m6A修饰图谱及修饰特征分析,不同实验分组间差异m6A修饰位点和基因鉴定;
√与RNA-seq、CLIP-seq数据等联合分析,揭示m6A修饰的功能。
meRIP-seq的应用案例
案例一—— 通过对FTO表达水平存在差异的HeLa细胞进行m6A-seq,并进行了FTO的CLIP-seq,通过整合分析m6A-seq数据和CLIP-seq数据,发现FTO过表达以浓度依赖性的方式特异性地从GGACU和RRACU基序中去除m6A修饰(Li et al. 2019)。
案例二——通过对强毒性和弱毒性的鸭甲肝病毒(DHAV)感染的雏鸭肝脏组织进行m6A-seq测序,首次分析了鸭子mRNA上的m6A修饰水平及分布特征,发现GAAGAAG是修饰基序最丰富的基序,整合分析m6A-seq和RNA-seq数据,发现m6A与DHAV感染雏鸭肝脏中mRNA表达水平总体呈正相关(Wu et al. 2022)。
参考文献
1.Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S. et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485, 201-206.
2.Li, Y., Wu, K., Quan, W., Yu, L., Chen, S., Cheng, C. et al. (2019). The dynamics of FTO binding and demethylation from the m(6)A motifs. RNA Biol, 16, 1179-1189.
3.Wu, L., Quan, W., Zhang, Y., Wang, M., Ou, X., Mao, S. et al. (2022). Attenuated Duck Hepatitis A Virus Infection Is Associated With High mRNA Maintenance in Duckling Liver via m6A Modification. Front Immunol, 13, 839677.
4.Yang, Y., Hsu, P.J., Chen, Y.-S. & Yang, Y.-G. (2018). Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res.