CLIP-seq技术介绍
CLIP-seq,又称为HITS-CLIP,即紫外交联免疫沉淀结合高通量测序(crosslinking-immunoprecipitation and high-throughput sequencing),是一项在全基因组水平揭示RNA分子与RNA结合蛋白相互作用的革命性技术。CLIP-seq实验是目前在全基因组水平上确定RNA结合蛋白(RBPs)结合位点的最重要手段(1, 2)。主要原理是基于RNA分子与RBP在紫外照射下发生共价结合,提高RNA结合蛋白与相应RNA靶标的结合强度;并通过蛋白免疫沉淀方法获得目标RBP的结合RNA片段,再通过高通量测序的方法,对结合RNA片段进行测序。对测序获得的CLIP-seq数据,需要通过生物信息学方法对数据进行计算分析。
产品优势
1. 准确性高:从活细胞交联开始,反应了体内环境下真实的分子间互作。
2. 特异性强:紫外辐射不会造成蛋白和蛋白之间的交联,能够鉴定靶蛋白和RNA 之间的直接相互作用。
3. 应用范围广:特别适用于研究剪接因子RNA结合图谱、miRNA作用靶点等研究
2. 特异性强:紫外辐射不会造成蛋白和蛋白之间的交联,能够鉴定靶蛋白和RNA 之间的直接相互作用。
3. 应用范围广:特别适用于研究剪接因子RNA结合图谱、miRNA作用靶点等研究
CLIP-seq技术适用范围
各种疾病的靶标、RNA结合蛋白、RBP-RNA复合物、circRNA-microRNA 相互作用
CLIP-seq技术实验流程
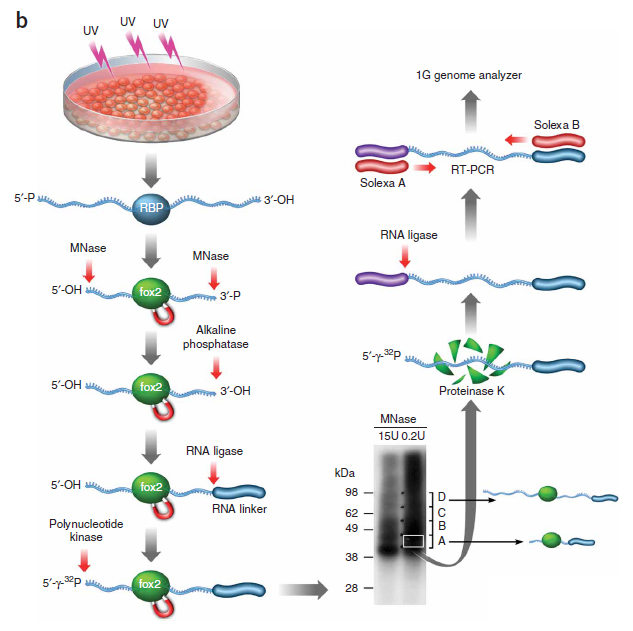
CLIP-seq交联和建库流程图

CLIP-seq建库路线图


参考文献
1. Uhl, M., et al., Computational analysis of CLIP-seq data. Methods, 2017. 118-119: p. 60-72.
2. Heyl, F., et al., Galaxy CLIP-Explorer: a web server for CLIP-Seq data analysis. GigaScience, 2020. 9(11).
2. Heyl, F., et al., Galaxy CLIP-Explorer: a web server for CLIP-Seq data analysis. GigaScience, 2020. 9(11).
案例解析
Hfq是一种普遍存在于细菌中的Sm-like RNA结合蛋白,与细菌的生理适应性和发病机制有关,但其在体内的结合特性仍不清楚。我们利用交联免疫共沉淀结合深度测序(CLIP-seq)的方法,在鼠疫耶尔森菌(鼠疫菌)中报道了全基因组hfq结合的RNA。(摘于文献摘要:Hfq Globally Binds and Destabilizes sRNAs and mRNAs in Yersinia pestis - PubMed (nih.gov)
