
近日,上海交通大学团队在《Nature Aging》发表重磅研究,首次揭示过氧化物酶体多功能酶 2(MFE-2)在 AD 病理中的关键作用,发现小分子化合物 CKBA 可通过靶向 MFE-2,重塑小胶质细胞脂质代谢与免疫稳态,为 AD 治疗提供全新靶点与候选药物。
中文题目:MFE-2 的缺失会损害小胶质细胞脂质稳态并驱动阿尔茨海默病发病机制中的神经炎症
发表期刊:Nature Aging(Q1 IF=19.4)
发表时间:2025.10
DOI 号:10.1038/s43587-025-00976-1
研究背景
近年来,研究发现在衰老和神经退行性病变的大脑中,小胶质细胞会出现脂滴积累。这种脂质代谢紊乱会促进活性氧(ROS)和促炎细胞因子的过量产生,从而加剧神经炎症和神经元损伤。然而,小胶质细胞中脂滴积累的起因及其如何影响AD中的小胶质细胞功能,仍是一个未解之谜。
过氧化物酶体是细胞进行脂肪酸β-氧化的关键场所之一,特别负责超长链脂肪酸(VLCFAs) 的降解。该过程对于维持中枢神经系统发育和稳态至关重要。多功能酶2(MFE-2) 是过氧化物酶体β-氧化通路中的核心酶,其基因(HSD17B4)突变在人类中会导致严重的神经发育障碍。尽管有零星证据提示MFE-2可能调节小胶质细胞功能,但其在AD pathogenesis中的具体角色和机制完全未知。
研究思路
- 靶点发现与临床关联:通过分析AD患者及模型小鼠的脑组织样本和公共数据库,确认MFE-2在病变小胶质细胞中的表达下调,确立其与AD病理的相关性。
- 体内功能验证:构建小胶质细胞特异性MFE-2敲除AD小鼠模型,通过行为学、免疫荧光、电镜及流式细胞术等技术,系统性验证MFE-2缺失会加剧小胶质细胞脂滴积累、Aβ沉积、线粒体功能障碍和认知缺陷。
- 分子机制解析:利用scRNA-seq、代谢组学、Seahorse能量代谢分析等技术,深入阐释MFE-2缺陷通过破坏过氧化物酶体-线粒体代谢轴,导致花生四烯酸累积和线粒体ROS过量产生,从而驱动促炎表型的分子机制。
- 治疗转化探索:聚焦转化医学,通过化合物筛选发现小分子CKBA能直接结合并稳定MFE-2,并在细胞和动物模型中证实其能有效逆转脂代谢异常、抑制神经炎症并改善AD病理,最终确立MFE-2为AD治疗的潜在新靶点。
主要研究结果
- 确认MFE-2是AD中小胶质细胞的关键失调分子
本研究首先发现在AD患者死后脑组织以及5xFAD模型小鼠的脑内,活化的小胶质细胞,尤其是Aβ斑块周边的小胶质细胞,其MFE-2的表达水平显著下调。结合对公共数据库的分析,证实MFE-2下调与AD关键风险基因APOE4相关联,确立了MFE-2缺失是AD中小胶质细胞的一个重要病理特征。
- 揭示MFE-2缺失通过破坏脂质代谢驱动神经炎症
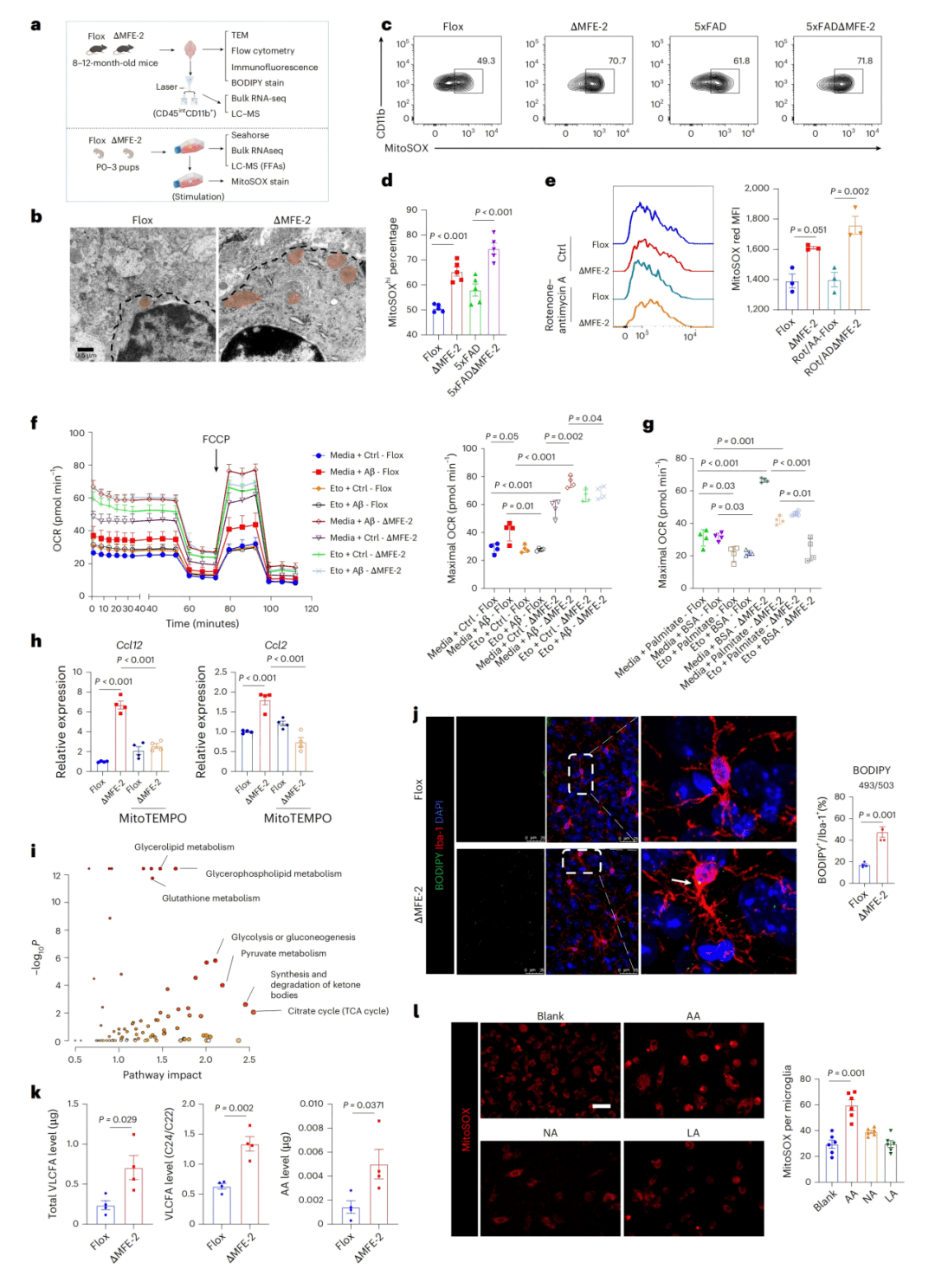
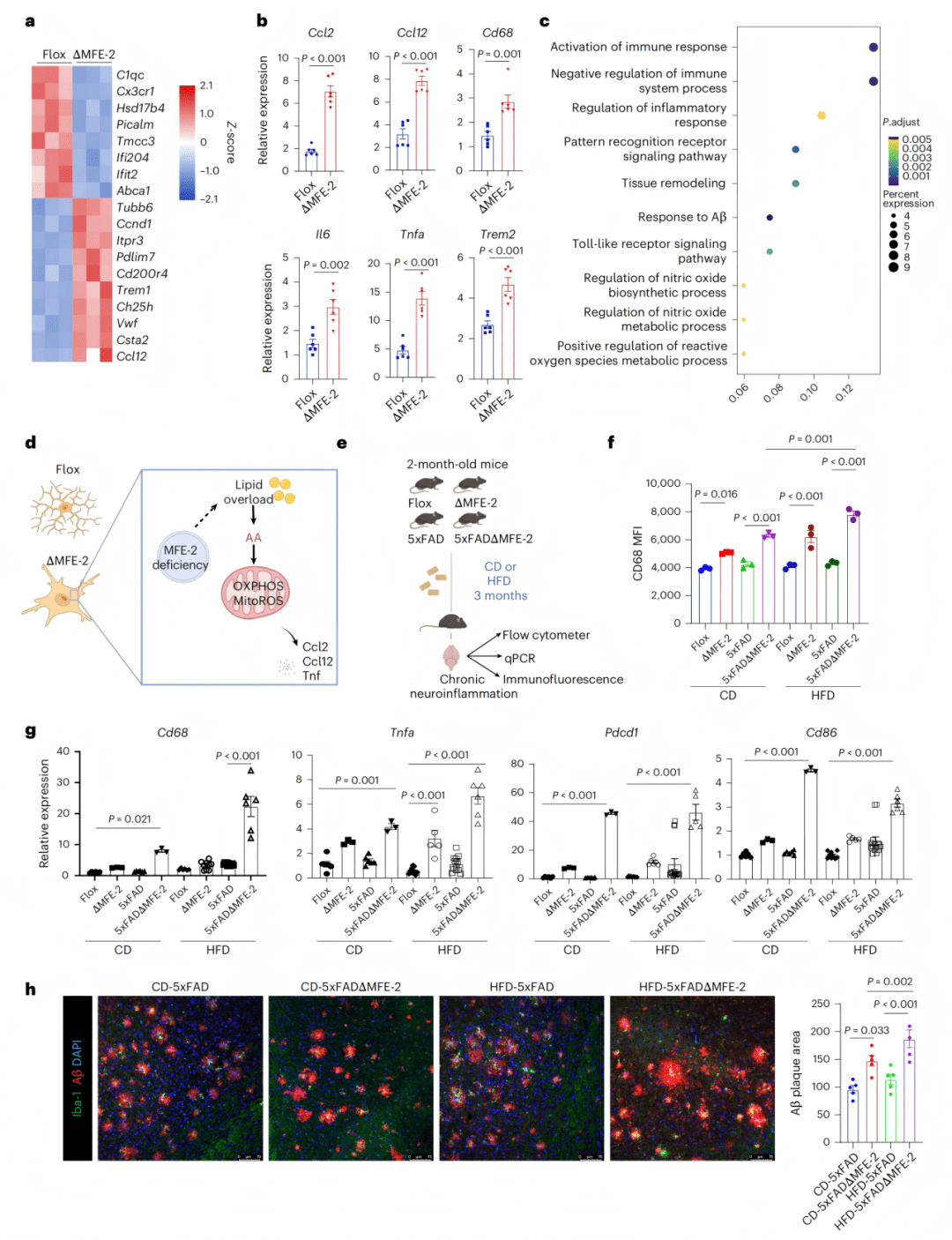
机制上,研究证实小胶质细胞特异性敲除MFE-2会导致过氧化物酶体β-氧化功能严重受损,引起极长链脂肪酸(VLCFAs)和花生四烯酸(AA)的显著累积。脂质堆积进一步诱发了线粒体功能紊乱,表现为氧化磷酸化基因上调、耗氧率异常增高以及线粒体活性氧(mtROS)的过量产生。这种代谢危机最终将小胶质细胞推向一种持续性的促炎状态,高表达CD68、IL-6、TNF等炎症因子,并上调Ccl2/Ccl12等关键趋化因子。
- 证明MFE-2缺陷直接加剧AD病理进程
在功能层面,小胶质细胞中MFE-2的缺失会削弱其吞噬Aβ的能力,导致脑中Aβ斑块负荷显著增加。同时,这些功能失调的细胞表现出疾病相关小胶质细胞(DAM)样表型,并伴随细胞衰老迹象。在动物行为学上,MFE-2敲除的AD模型小鼠表现出更严重的记忆缺陷和探索行为减少,直接证明了MFE-2缺陷在体内会加速AD的神经退行性进程。
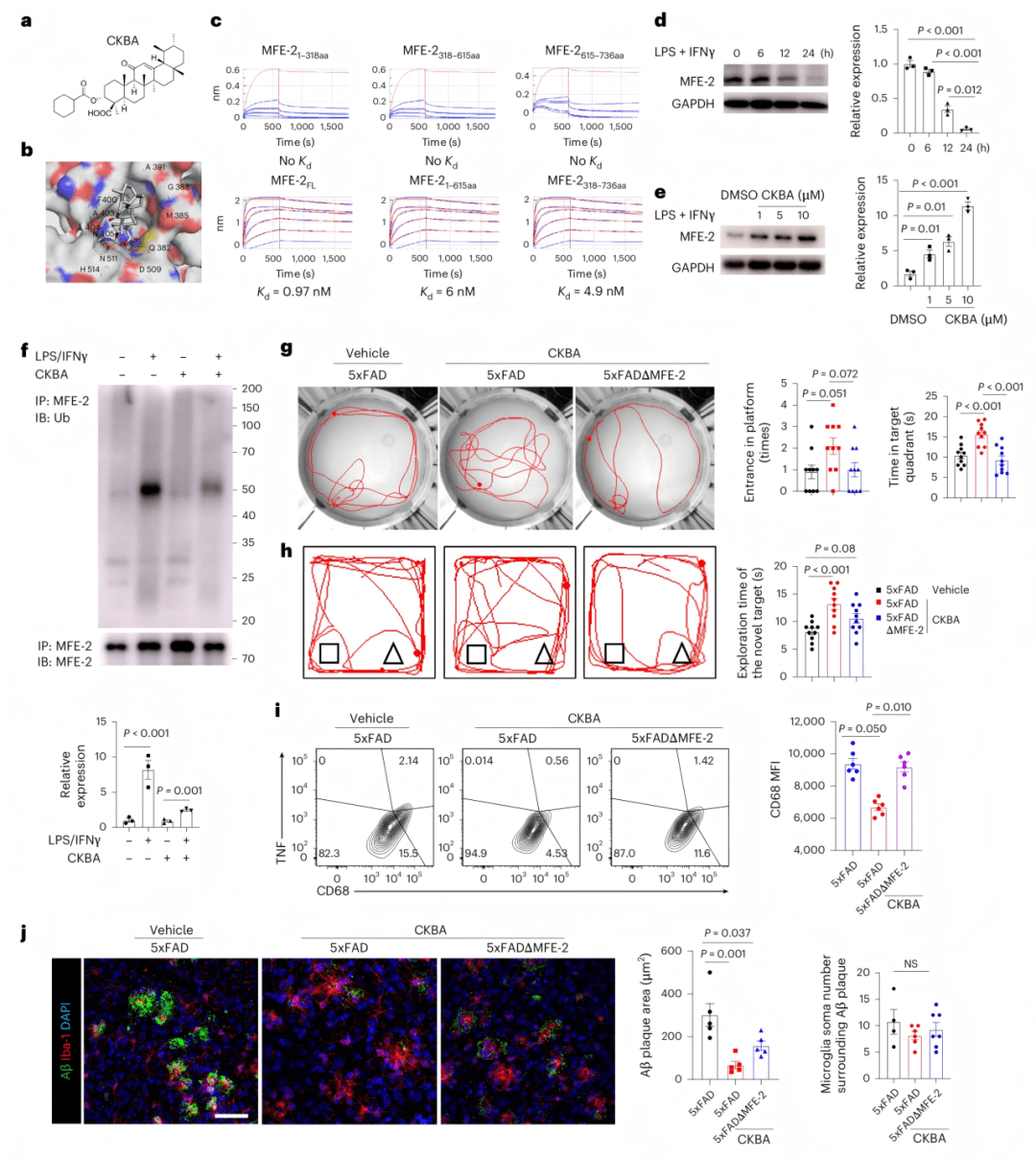
- 发现靶向MFE-2的小分子化合物CKBA具有治疗潜力
转化研究方面,团队筛选并鉴定出天然产物衍生物CKBA是MFE-2的直接配体,它能结合MFE-2并抑制其被泛素化降解,从而在炎症环境下稳定MFE-2蛋白水平。在AD模型小鼠中,CKBA治疗能有效降低小胶质细胞活化水平、减少mtROS、减轻Aβ病理,并显著改善小鼠的学习记忆功能。值得注意的是,CKBA的治疗效果在MFE-2敲除小鼠中被显著削弱,证实了其作用依赖于MFE-2靶点。
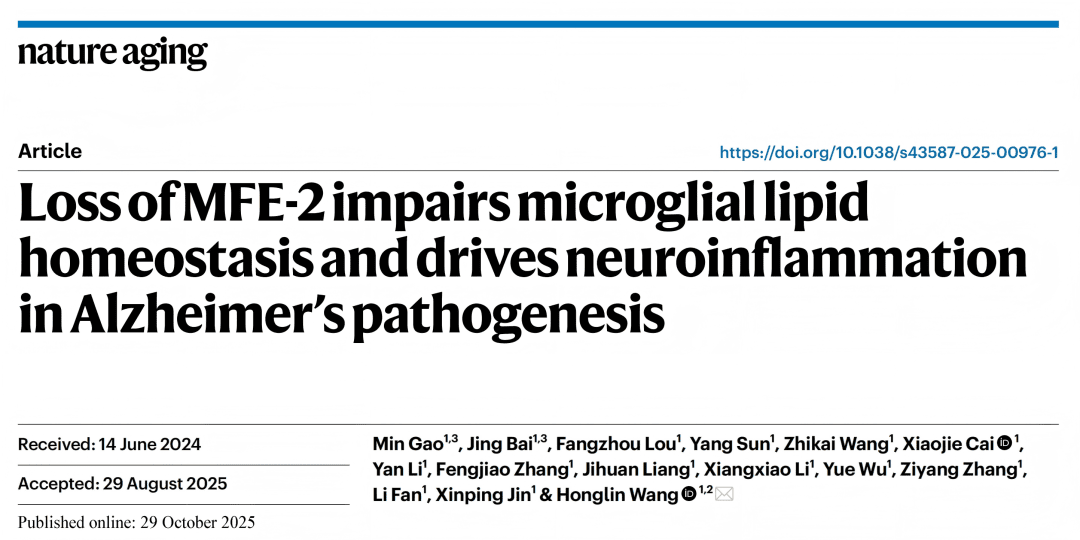
● 1a-c:转录组分析显示脂滴富集微glia中β-氧化基因上调,但APOE4微glia中MFE-2表达降低。
● 1d-e:免疫荧光证实人与AD小鼠脑中MFE-2蛋白水平下降。
● 1f:MFE-2敲除增加Aβ斑块负荷。
● 1g-i:水迷宫及旷场实验显示MFE-2缺失导致学习记忆能力下降。
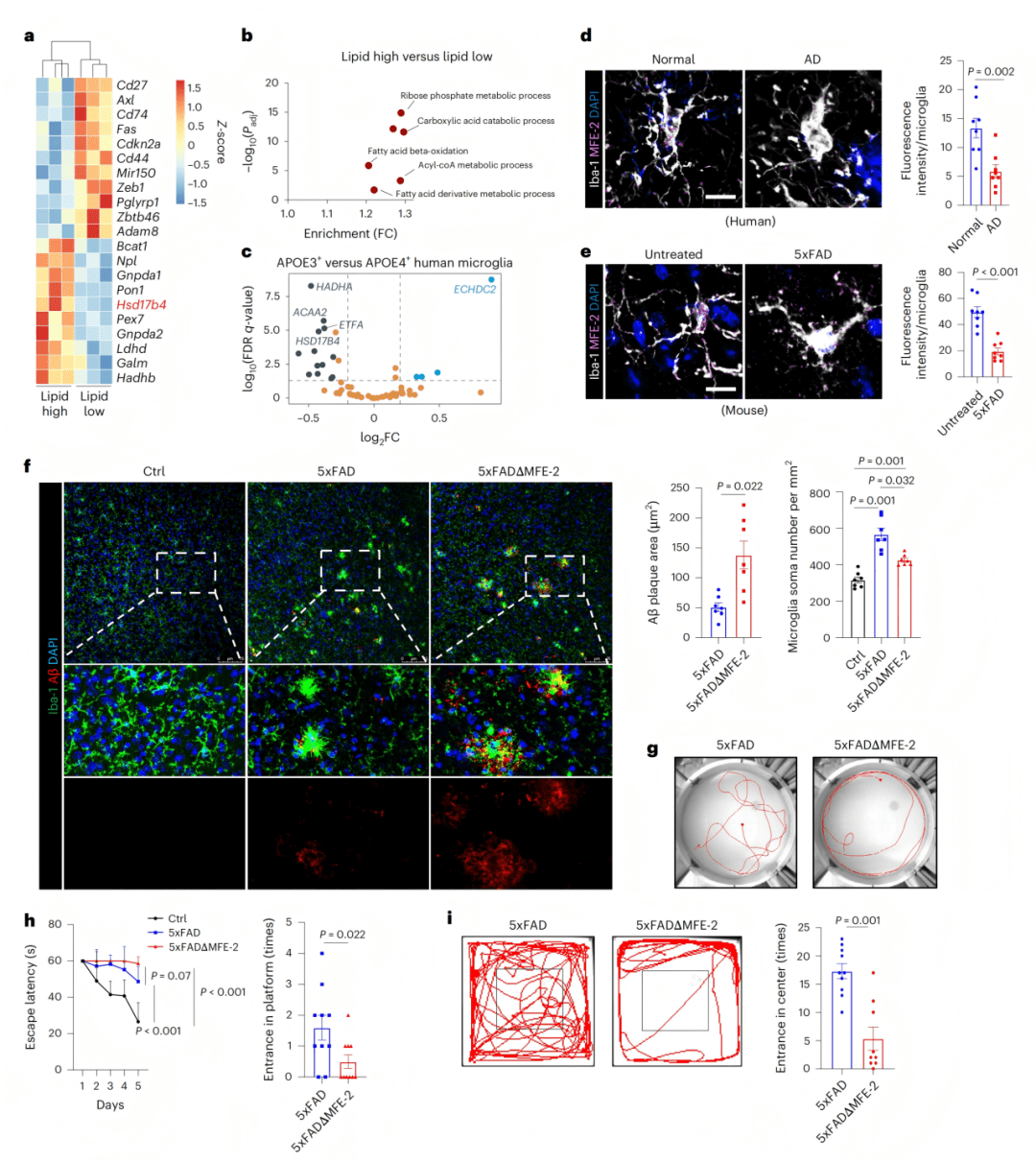
● 2a-b:scRNA-seq显示MFE-2缺失微glia聚类改变。
● 2c:OXPHOS相关基因表达上升。
● 2d-e:促炎基因富集,GO分析提示炎症通路激活。
● 2f-h:微glia形态变粗、SA-β-gal阳性率升高、CD68表达增加。
● 2i:批量RNA-seq显示IL-6通路激活。
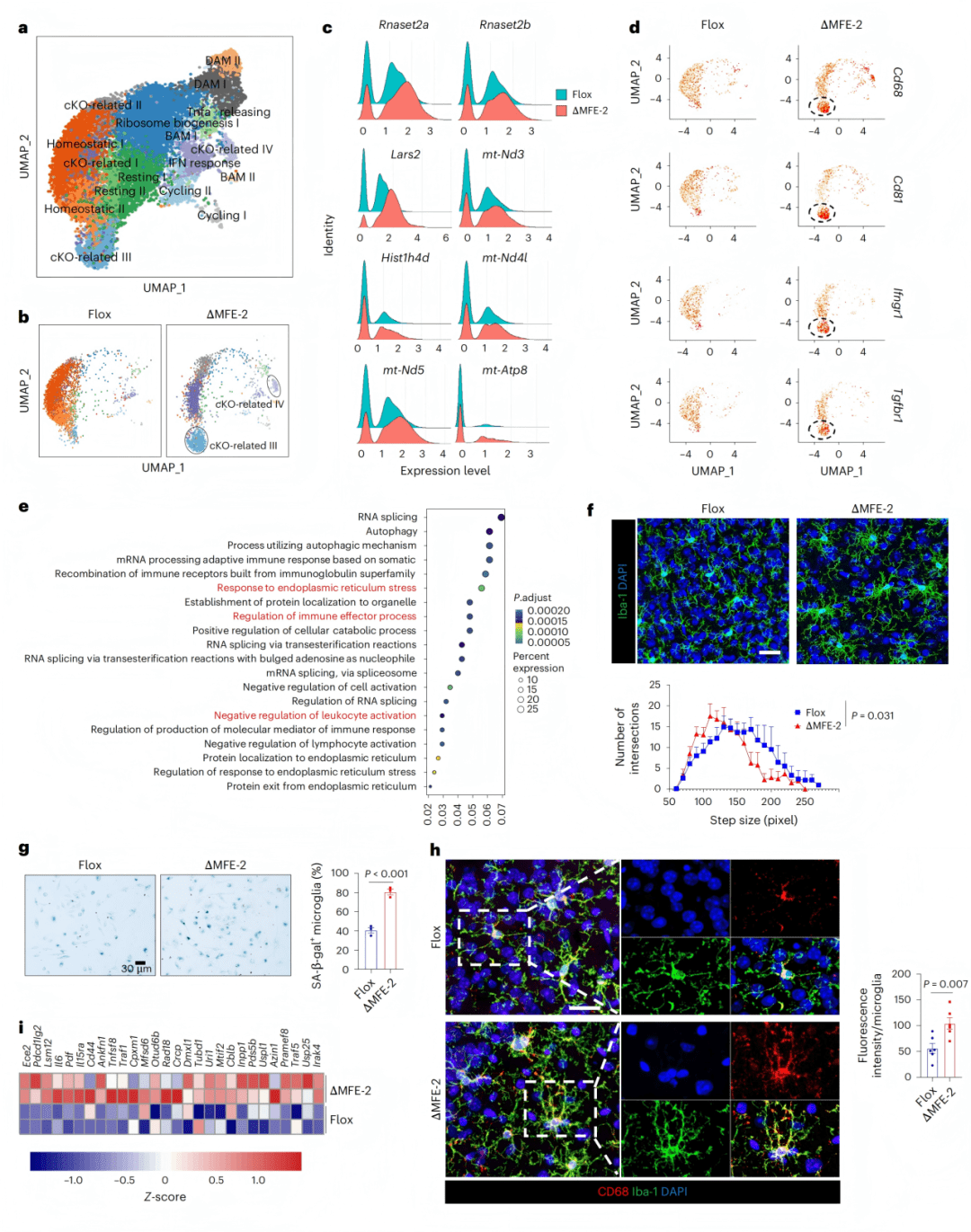
● 3a-c:DAM集群在MFE-2缺失脑中增多,CD11c免疫染色验证。
● 3d-f:qPCR和流式细胞术显示Il6、Cd68、IL-6R表达升高。
● 3g:MFE-2依赖的微glia亚群特征总结。
● 3h-j:Aβ吞噬实验显示MFE-2缺失微glia吞噬能力下降,Aβ刺激后炎症反应更强。
● 4a-b:实验示意图与TEM显示线粒体结构异常。
● 4c-e:MitoSOX检测显示基础及诱导后ROS均升高。
● 4f-g:Seahorse分析显示脂肪酸氧化代谢亢进。
● 4h:mitoTEMPO可抑制Ccl2/Ccl12表达。
● 4i-l:代谢组学显示花生四烯酸积累,且外源AA可诱导ROS。
● 5a-b:RNA-seq与qPCR显示Ccl2/Ccl12显著上调。
● 5c:GO分析显示免疫与氧化应激通路富集。
● 5d:机制示意图。
● 5e-h:高脂饮食增强CD68表达、促炎基因水平及Aβ斑块沉积。
● 6a-c:CKBA结构与结合验证(Kd=0.97 nM)。
● 6d-f:CKBA逆转炎症导致的MFE-2下调并减少其泛素化。
● 6g-h:行为学测试显示CKBA改善记忆与探索行为。
● 6i-j:CKBA降低CD68表达及Aβ斑块负荷。
Gao M, Bai J, Lou F, et al. Loss of MFE-2 impairs microglial lipid homeostasis and drives neuroinflammation in Alzheimer’s pathogenesis. Nat Aging. Published online October 29, 2025. doi:10.1038/s43587-025-00976-1