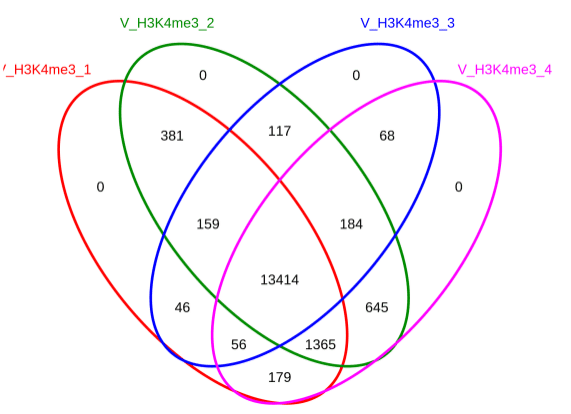
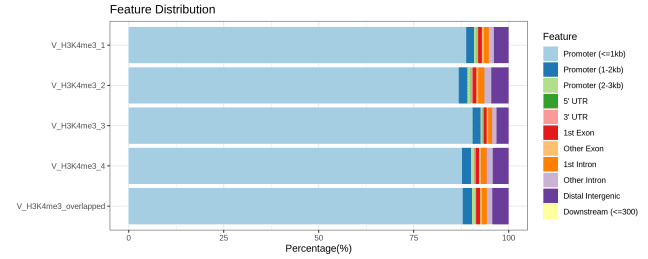
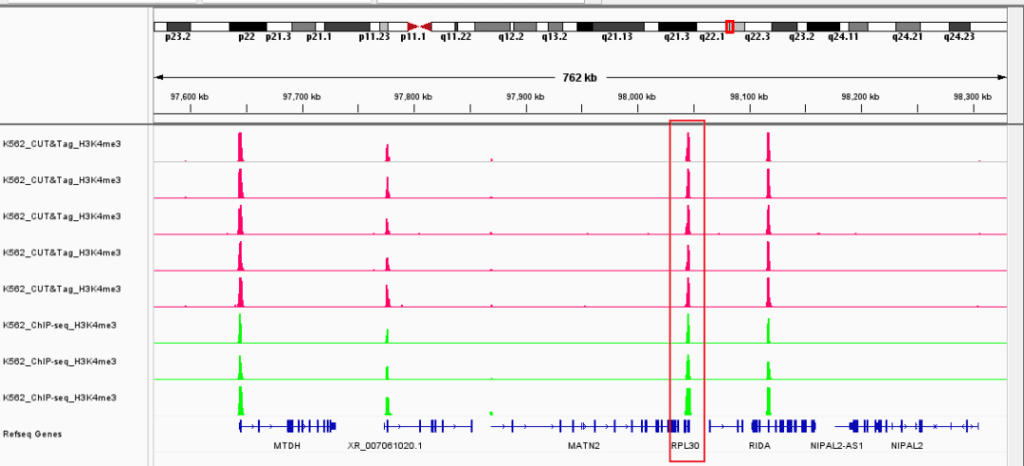
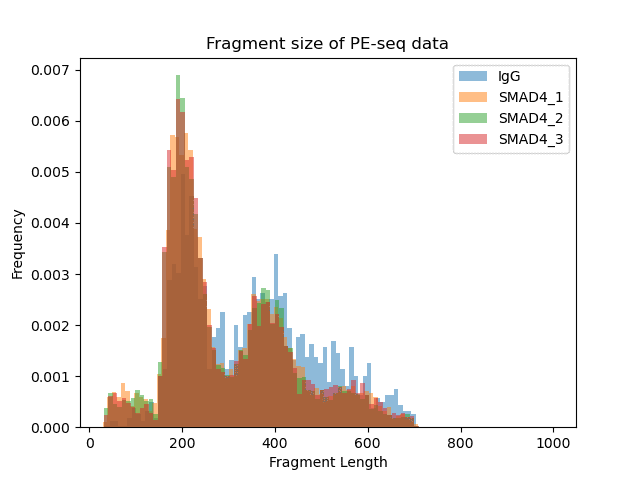
RXBio Translates Sequence to Science and Industry

2019年4月29日,Henikoff实验室在Nature Communication上公布了一项革命性的新技术,名为CUT&Tag(Cleavage Under Targets and Tagmentation)。这一创新技术彻底改变了ChIP-seq实验的细胞量需求,使其从原先的10,000个大幅降低到了60个,甚至单个细胞,这无疑是对生物学研究领域的一次重大突破。CUT&Tag技术的出现,不仅极大地提高了实验的可行性和效率,更在传统ChIP-seq实验中存在的信噪比(信号与噪声的比率)和数据重复性等问题方面实现了质的飞跃。
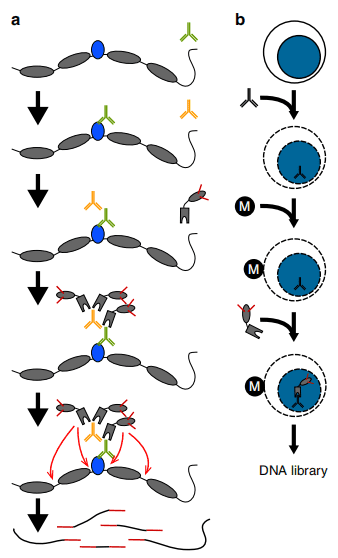
CUT&Tag技术是一种革新性的蛋白质-DNA互作研究方法,无需交联、超声打断、末端修复和接头连接等操作,因此具有所需样本量少、实验周期更快、信噪比高和可重复性好等优势,甚至可用于单细胞水平测序,尤其适用于早期胚胎发育、干细胞、肿瘤以及表观遗传学等研究领域。CUT&Tag有望将蛋白质-DNA互作的研究变成一种类似PCR反应的常规操作,对基因调控、表观遗传等领域的研究具有革命性的意义。
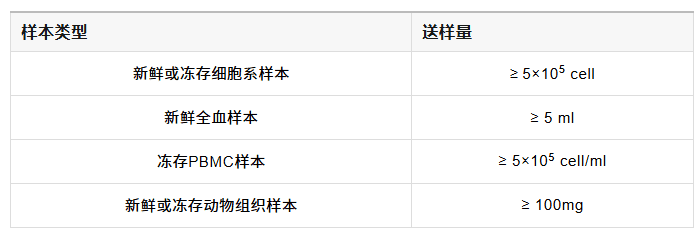
✔ 起始量低:所需要细胞数100-100,000个细胞,甚至可以做单细胞水平研究;
✔ 流程简单:无需甲醛交联、超声打断、免疫共沉淀,末端补平,加A和接头;
✔ 特异性强:更高的信噪比和更低的背景;
✔ 性价比高:所需测序深度少;
✔ 重复性好:实验重复结果一致性好。
该技术不需要使用免疫共沉淀,而是通过针对靶蛋白的抗体和Protein A的介导,成功实现了Tn5酶(已与Protein A融合)在切割DNA片段的同时,在序列两端添加测序接头。经PCR扩增后,可以直接生成用于高通量测序的文库。该技术的核心步骤为“切割下目标并进行扩增”(Cleavage Under Targets and Tagmentation),这一步骤以前所未有的精度和灵敏度,针对极少量细胞进行了基因组范围内的蛋白质-DNA相互作用研究。使用CUT&Tag技术,可以大大降低细胞投入量以及操作时间,仅需一天即可获得高质量的高通量测序文库。
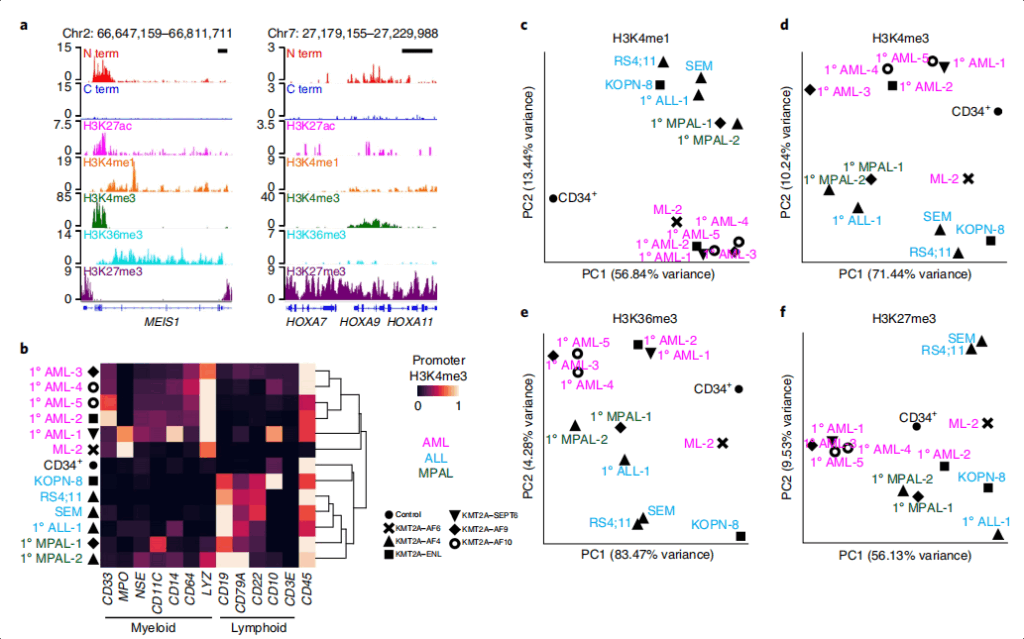
Steven Henikoff团队建立了AutoCUT&Tag平台,可全自动分析不同细胞系和患者样本中的肿瘤融合蛋白、转录相关复合物和组蛋白修饰,改进了CUT&Tag染色质分析技术。发现KMT2A肿瘤融合蛋白诱导的特异性位点,并发现不同的融合基因对各种转录辅助因子表现出不同的亲和力,可作为预测癌症对不同治疗性化合物敏感性的工具。研究成果已发表在Nature Genetics上,题为“Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia”。
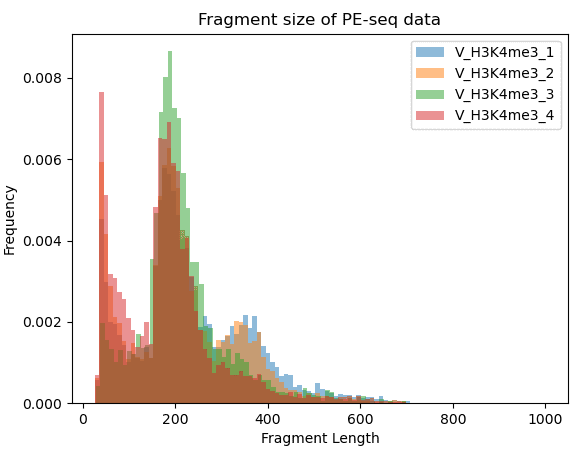
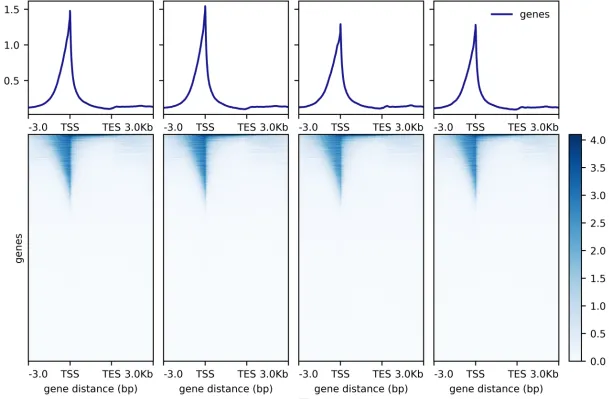
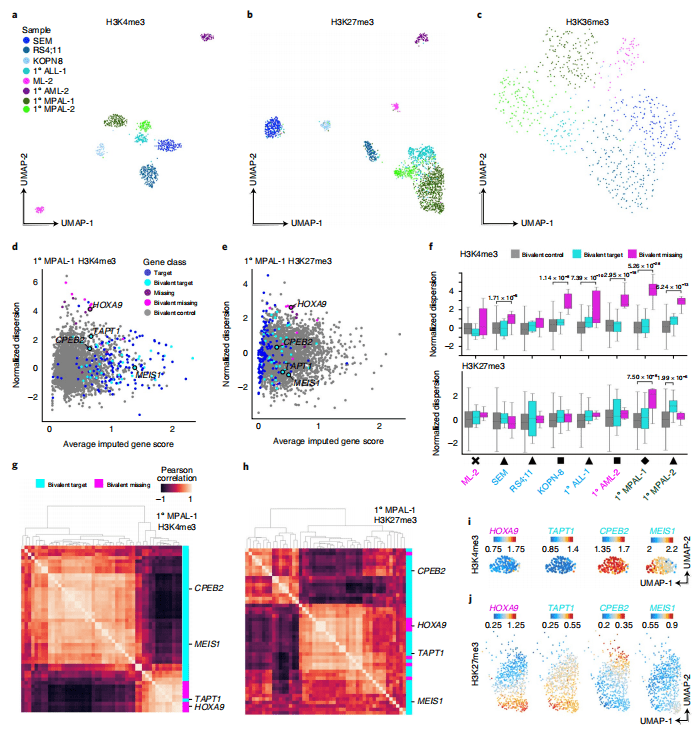
研究团队利用AutoCUT&Tag分析白血病样本中的组蛋白修饰,发现了具有二价染色质特征的KMT2A肿瘤融合蛋白位点。KMT2A肿瘤融合结合位点的一个子集被二价(H3K4me3和H3K27me3)染色质标记,并且这些位点上的二价染色质特征与同一肿瘤细胞之间的异质性相关,这表明混合谱系白血病细胞群体中基因表达的异质性源于染色质动力学。
对H3K4me3和H3K27me3的单细胞CUT&Tag分析,筛选出一组在大多数KMT2Ar白血病中共享的基因。这些基因在白血病细胞中起着关键作用,并且与肿瘤细胞的生长和分化密切相关。然而,在部分KMT2Ar白血病样本中,这些靶基因缺失,导致肿瘤内活性和抑制性染色质标记变化最大。这些缺失靶点可能是由于肿瘤内有限细胞亚群中的肿瘤蛋白结合、激活,从而导致其降低至KMT2A谱分析检测水平以下。这也表明了KMT2A融合蛋白对白血病细胞的生长和分化具有重要作用,而这种作用可能受到不同细胞亚群中基因表达异质性的影响。这一发现不仅有助于深入了解KMT2A融合蛋白在白血病中的作用,也为开发针对这种融合蛋白的靶向治疗提供了新的思路。在未来的研究中,可以进一步探讨这些基因异质性的机制以及如何利用这些信息来提高白血病的治疗效果。
参考文献:
1. Kaya-Okur, H.S., Wu, S.J., Codomo, C.A. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). https://doi.org/10.1038/s41467-019-09982-5
2. Janssens, D.H., Meers, M.P., Wu, S.J. et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat Genet 53, 1586–1596 (2021). https://doi.org/10.1038/s41588-021-00941-9

电话:027-870502099
邮箱:sales@rxbio.cc
地址:武汉市东湖高新区高新二路388 号
光谷生物医药加速器 18 栋 1-2层
单细胞多组学 空间转录组
三代测序 功能基因组
表观遗传学 互作组学
单细胞大数据 数据深度挖掘

欢迎关注公众号「瑞兴生物」