
食管鳞状细胞癌(ESCC)的代谢异常与治疗抵抗是临床难题。乳酸作为肿瘤代谢核心产物,其介导的蛋白质乳酰化修饰在DNA修复、免疫逃逸中已有报道,但能否调控选择性多聚腺苷酸化(APA)——这一影响70%基因3′ UTR长度与mRNA命运的关键机制——仍属未知。
中山大学肿瘤医院发表在 Cell Discovery 上的研究(Lin et al., 2025)通过临床数据→分子机制→动物模型三重验证,首次揭示了”乳酸代谢→NUDT21乳酰化→FDX1 3’UTR延长→铜死亡逃逸”的靶向通路,为ESCC靶向治疗提供新策略。
瑞兴生物提供的PAS-seq(多聚腺苷酸化位点测序)与RNA-seq技术助力本研究核心突破:精准解析乳酸诱导的APA重编程机制,锁定NUDT21乳酰化-FDX1调控轴,为铜死亡抵抗提供关键组学证据。
PAS-seq技术更多了解:全网最详细 | 一文读懂APA及其研究利器PAS-seq
01
原文概览

英文标题:NUDT21 lactylation reprograms alternative polyadenylation to promote cuproptosis resistance
中文标题:NUDT21乳酰化重编程选择性多聚腺苷酸化以诱导铜死亡抵抗
期刊:Cell Discovery
影响因子IF:12.5
发表时间:2025.3
DOI: 10.1038/s41421-025-00804-1
02
技术路线
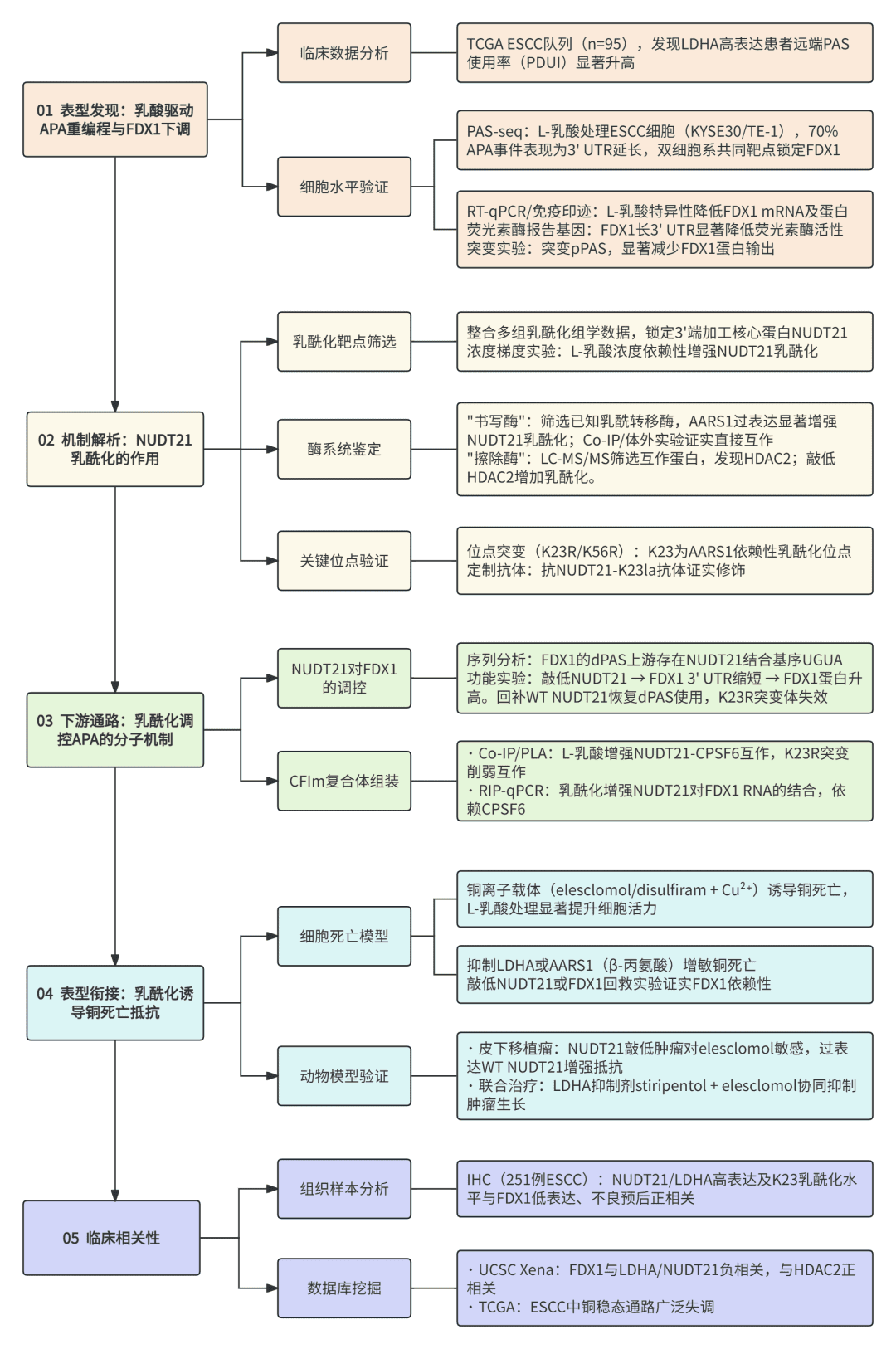
03
主要结果
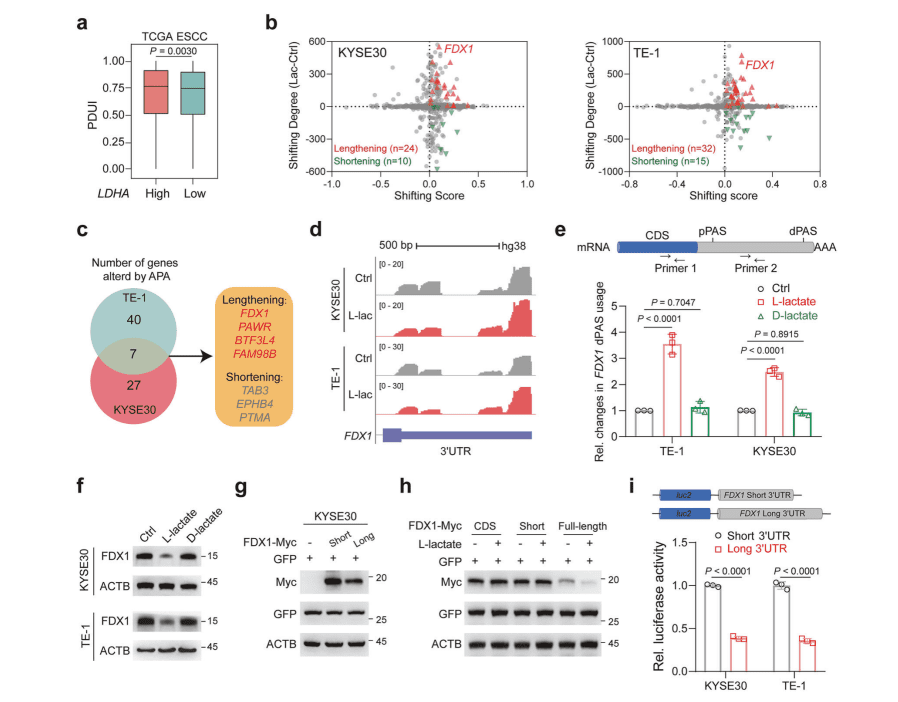
01 L-乳酸驱动食管鳞癌中的APA重编程
乳酸是多种生物过程的成熟调节剂,然而它在 APA 中的作用在很大程度上仍未得到探索。
▪ 研究团队分析TCGA食管鳞癌(ESCC)数据,发现高LDHA(负责乳酸产生的主要酶)表达患者表现出优先使用远端 PAS (dPAS);
▪ 通过PAS-seq和RT-qPCR检测L-乳酸处理的ESCC细胞(KYSE30/TE-1),验证FDX1、PAWR等基因的3′ UTR延长。
▪ 构建FDX1 3′ UTR长短变体载体,结合免疫印迹和双荧光素酶报告基因实验,明确长3′ UTR降低FDX1蛋白表达。
▪ 对比D-乳酸处理,证明调控具有L-乳酸特异性。
核心结论:L-乳酸(非D-乳酸)通过诱导FDX1等基因的3′ UTR延长,显著降低FDX1蛋白水平,且长3′ UTR直接削弱其翻译效率。
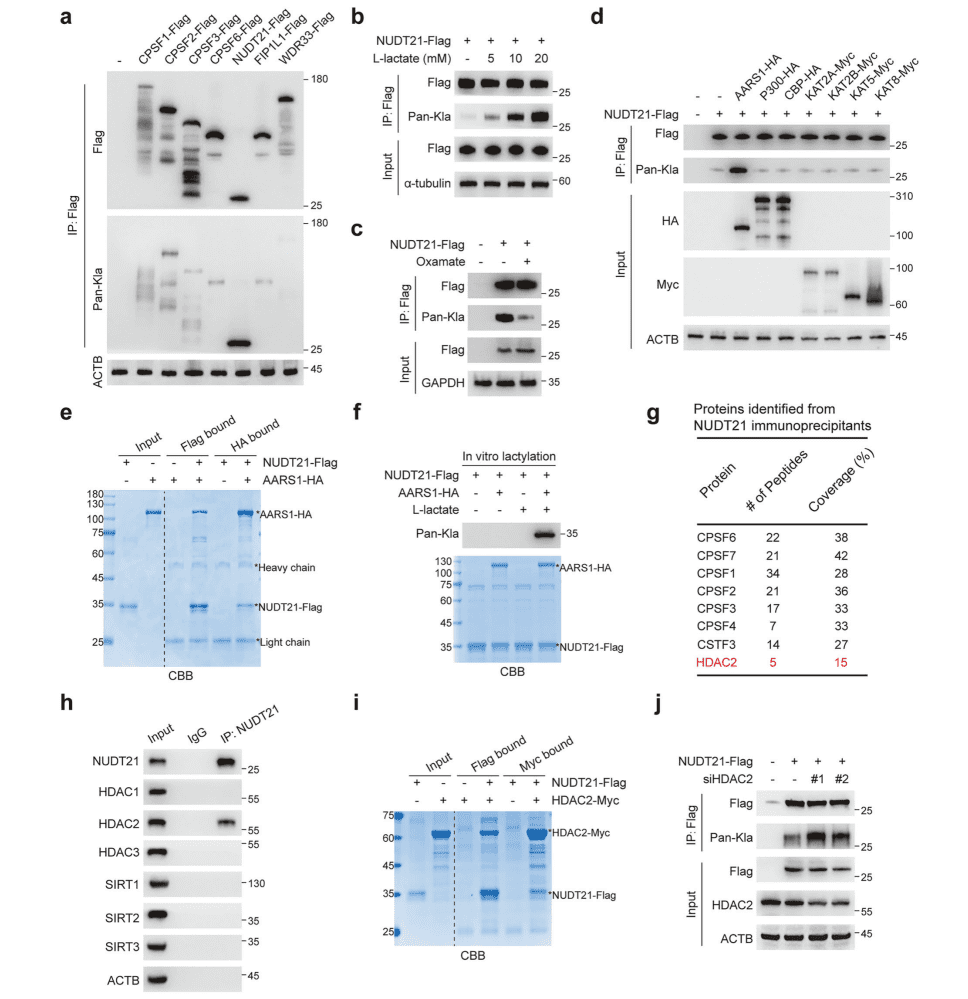
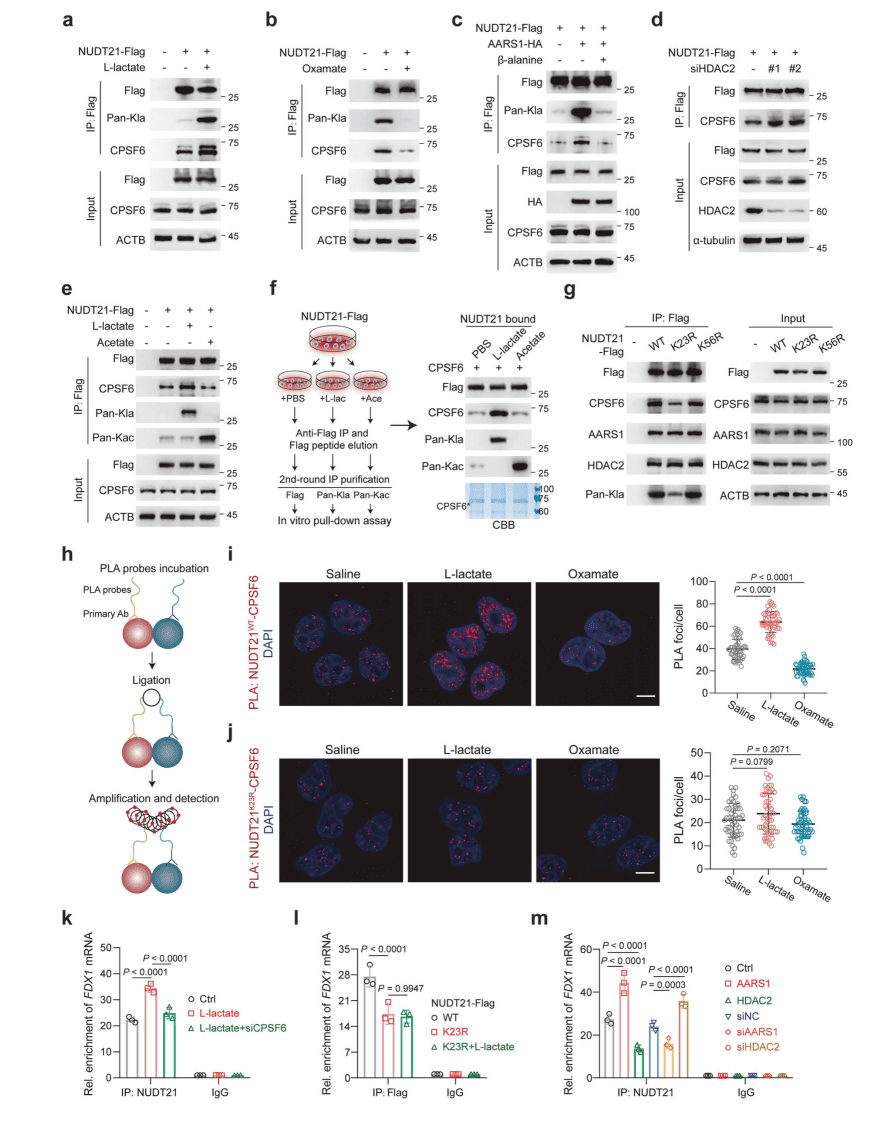
02 L-乳酸促进NUDT21乳酰化
蛋白质乳酰化是一种由L-乳酸介导的翻译后修饰,作为关键调控信号参与多种生物学过程的调节。为了研究 L-乳酸是否通过乳酰化介导 APA,研究团队:
▪ 整合乳酰化组学数据,筛选出3′ UTR加工核心蛋白NUDT21为乳酰化靶点。
▪ 浓度梯度L-乳酸处理及LDHA抑制剂(oxamate)验证NUDT21乳酰化水平变化。
▪ 通过Co-IP、体外互作和酶活实验,鉴定AARS1为乳酰转移酶、HDAC2为去乳酰酶。
核心结论:L-乳酸通过AARS1催化NUDT21乳酰化,HDAC2反向去乳酰化,形成动态修饰调控。
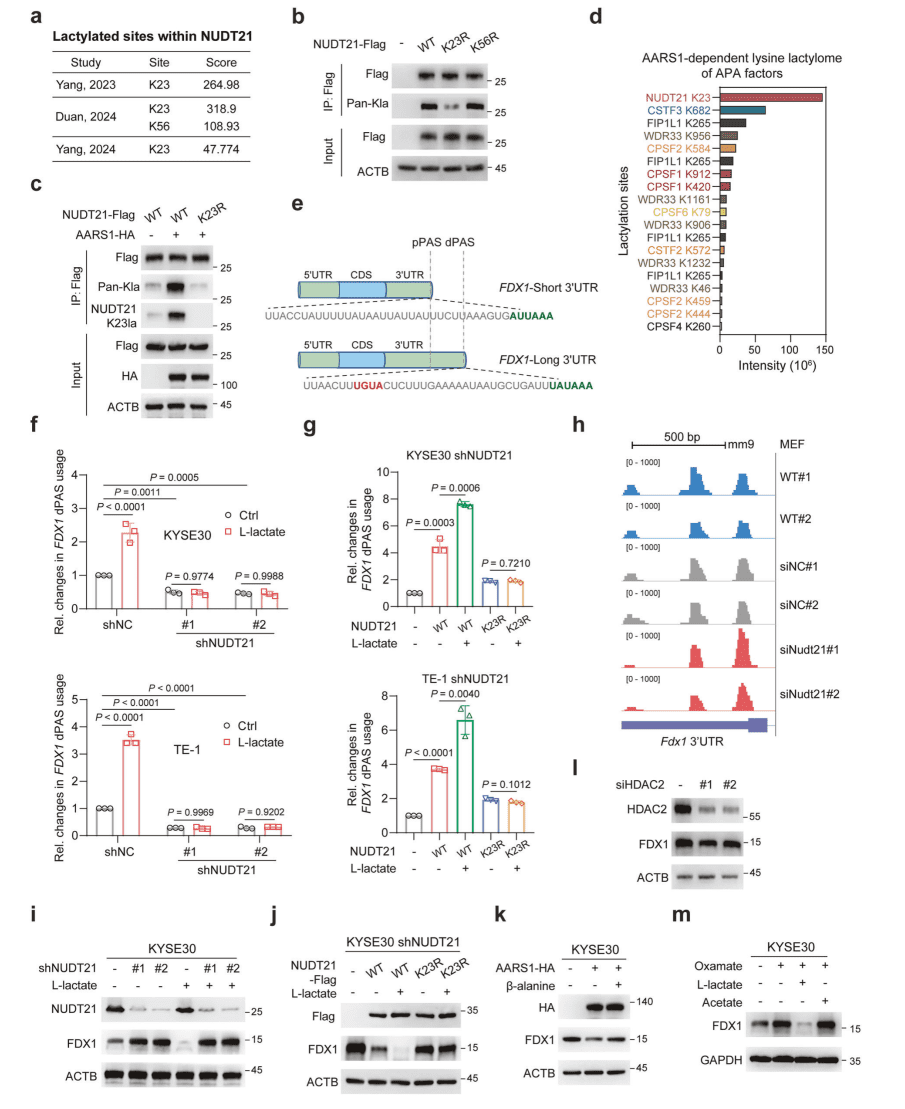
03 NUDT21 K23位点乳酰化促进
FDX1远端PAS的使用
先前的研究表明,3′ 末端加工机械的几个组件可以进行乳酰化,残基 K23 和 K56 被确定为 NUDT21 上的潜在乳酰化位点。研究团队:
▪ 质谱鉴定NUDT23乳酰化位点(K23为主),K23R突变体验证其关键性。
▪ NUDT21敲低后FDX1的3′ UTR缩短,回补WT(非K23R突变体)恢复dPAS使用。
▪ AARS1过表达/抑制剂(β-丙氨酸)、HDAC2敲低/过表达调控FDX1蛋白水平,依赖NUDT21乳酰化。
▪ 对比乙酸处理,排除乙酰化干扰,证实乳酰化特异性。
核心结论:NUDT21 K23乳酰化特异性促进FDX1远端PAS选择,导致3′ UTR延长及蛋白表达抑制。
04 NUDT21乳酰化促进CFIm复合体形成
形成 CFIm 复合物的 NUDT21 和 CPSF6 之间的相互作用代表了 APA 中的关键早期事件,促进了 3′ 末端加工机械的募集和组装,因此,团队研究了 NUDT21 乳酸是否调节 CFIm 复合物的形成。
▪ Co-IP和PLA实验显示L-乳酸增强NUDT21-CPSF6互作,oxamate/β-丙氨酸抑制互作。
▪ 体外pull-down证明乳酰化NUDT21(非乙酰化)优先结合CPSF6,K23R突变削弱互作。
▪ RIP-qPCR证实乳酰化提升NUDT21与FDX1 RNA结合,且依赖CPSF6。
核心结论:NUDT21 K23乳酰化通过增强CFIm复合体(NUDT21-CPSF6)组装,提升其对FDX1 RNA的结合能力。
05 NUDT21乳酰化通过
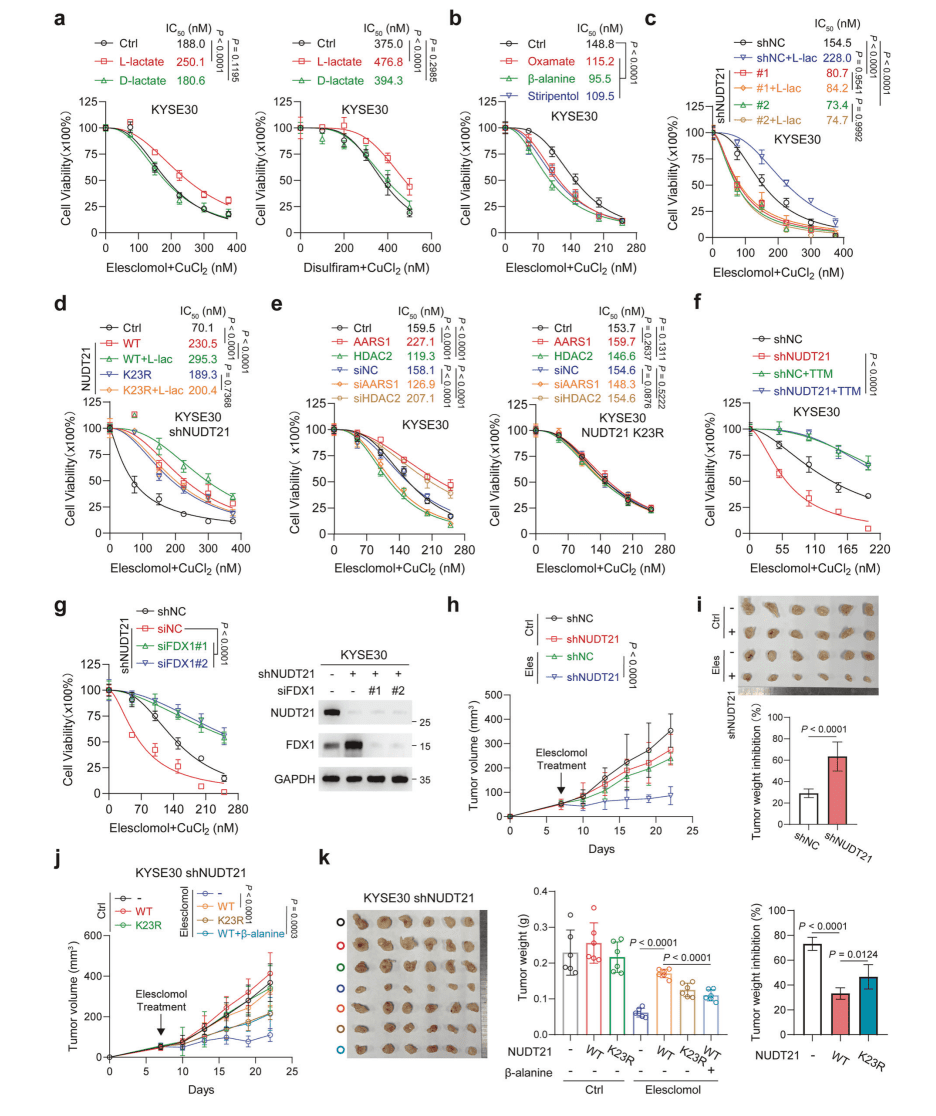
靶向FDX1赋予铜死亡抵抗性
鉴于 FDX1 在调节铜死亡中的核心作用,团队研究了乳酰化 NUDT21 是否会影响细胞对 ESCC 中铜诱导应激的敏感性。
▪ L-乳酸处理显著降低铜离子载体(elesclomol/disulfiram+Cu²⁺)诱导的细胞死亡,而oxamate/β-丙氨酸增敏。
▪ NUDT21敲低或K23R突变消除乳酸保护作用,铜螯合剂(TTM)可逆转死亡。
▪ 体内实验:NUDT21敲低肿瘤对elesclomol敏感,K23R突变或β-丙氨酸处理抑制肿瘤生长。
▪ FDX1敲除逆转NUDT21缺失导致的铜死亡增敏。
核心结论:NUDT21乳酰化通过下调FDX1介导ESCC铜死亡抗性,靶向该轴(如β-丙氨酸+elesclomol)可协同抑制肿瘤。
06 乳酸-NUDT21-FDX1-铜死亡轴
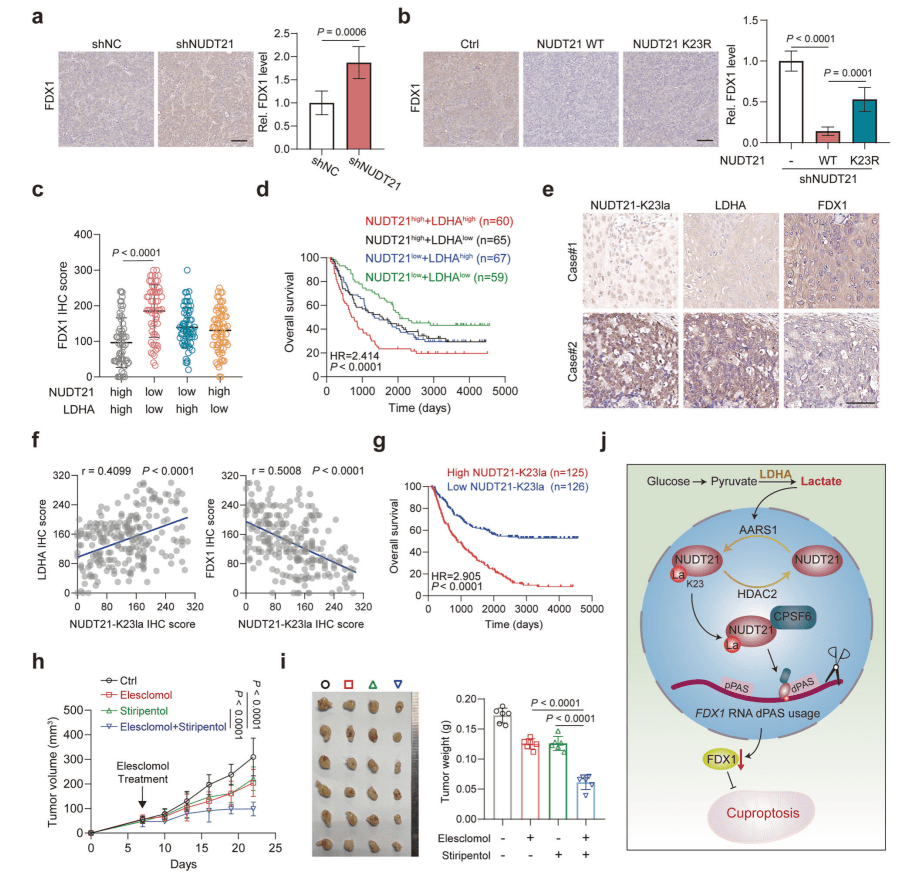
是食管鳞癌治疗的潜在靶点
▪ 分析TCGA ESCC转录组数据,揭示FDX1表达与NUDT21、LDHA、AARS1呈显著负相关,铜代谢通路基因(如铜转运蛋白)及金属硫蛋白(MTs)在ESCC中广泛失调。
▪ 基于251例ESCC临床样本的免疫组化(IHC),量化K23-乳酰化NUDT21、LDHA、FDX1蛋白水平,验证其表达相关性(如高LDHA/NUDT21组FDX1↓,生存率↓)。
▪ 生存分析显示高K23-乳酰化NUDT21患者预后最差(P<0.001),且与FDX1低表达强负相关(r=-0.68)。
▪ 小鼠移植瘤模型:ESCC细胞(KYSE30)移植后,联合使用临床LDHA抑制剂stiripentol和铜离子载体elesclomol,显著抑制肿瘤生长(vs. 单药,P<0.01)。
▪ IHC验证体内机制:NUDT21敲低肿瘤中FDX1蛋白↑,而回补WT NUDT21(非K23R突变)后FDX1↓,且联合治疗组肿瘤铜含量不变(排除铜代谢干扰)。
核心结论:
a.临床意义:ESCC中乳酸-NUDT21-FDX1轴驱动铜死亡抗性——高LDHA/NUDT21及K23-乳酰化NUDT21水平直接导致FDX1表达抑制,且与患者不良预后显著相关(HR=2.91, P<0.0001)。
b.治疗策略:靶向该轴的联合疗法(stiripentol抑制LDHA + elesclomol诱导铜死亡)通过阻断乳酸代谢→恢复FDX1表达→重启铜死亡,在小鼠模型中实现协同抑瘤(肿瘤体积↓68%,P<0.001),且无显著毒性,为ESCC提供临床转化潜力。
04
研究总结
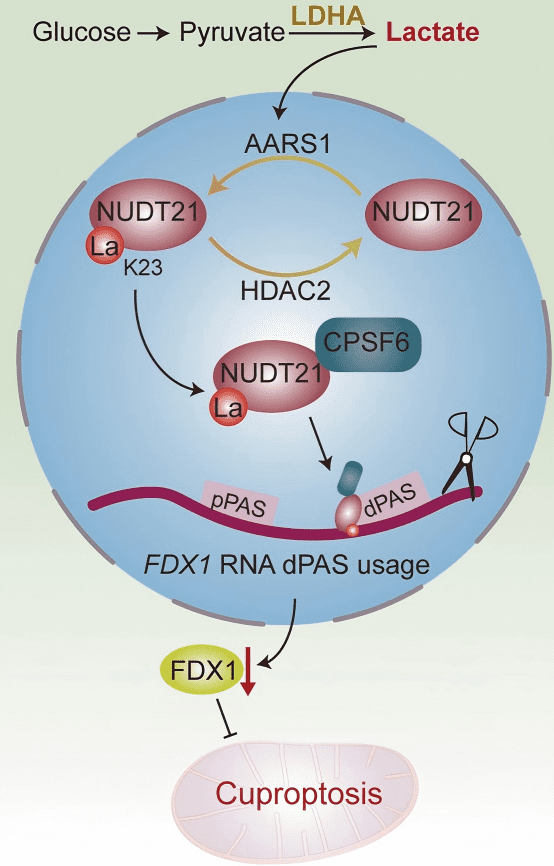
L-乳酸 → AARS1介导NUDT21 K23乳酰化 → 增强CFIm复合体形成 → FDX1基因3′ UTR延长 → FDX1蛋白下调 → 铜死亡抵抗 → ESCC进展
靶向策略:
临床LDHA抑制剂stiripentol阻断乳酸生成 → 抑制NUDT21乳酰化 → 恢复FDX1表达 → 增强铜离子载体elesclomol的铜死亡诱导作用 → 协同抑制ESCC
参考文献
Lin J, Yin Y, Cao J, et al. NUDT21 lactylation reprograms alternative polyadenylation to promote cuproptosis resistance. Cell Discov. 2025;11(1):52. Published 2025 May 28. doi:10.1038/s41421-025-00804-1