
Introduction
In 1958, Francis Crick, the discoverer of the DNA double helix structure, put forward the “Central Dogma”, which elaborated on the information flow among DNA, RNA, and proteins. Gene transcription generates precursor RNA, which undergoes alternative splicing to form multiple transcripts, and these transcripts are then translated into different protein isoforms. Alternative splicing enables genes to produce multiple protein isoforms, which may have significant differences in structure and function and are specifically expressed in specific diseases, tissues, or developmental stages. Researchers have discussed the role of protein isoforms in drug discovery and how to utilize them to enhance drug specificity and therapeutic effects in the journal “Nature Reviews Drug Discovery” [1]. Therefore, studying the regulatory mechanisms of alternative splicing of precursor RNA and how these transcripts are translated into proteins and perform their functions is crucial for the development of treatments centered around protein isoforms.
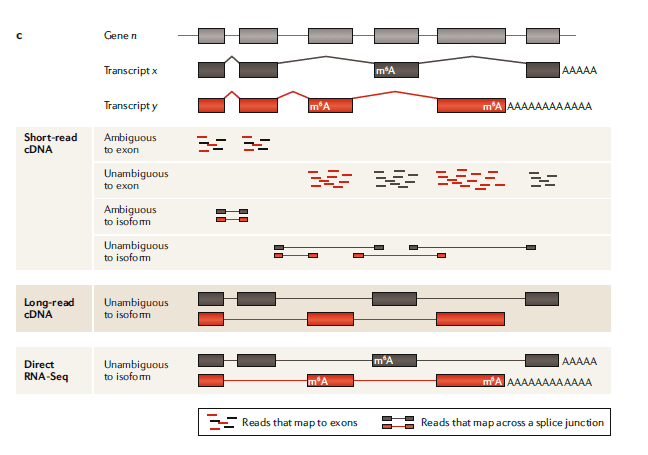
In the human genome, more than 90% of genes undergo alternative splicing to produce multiple transcripts. In short-read cDNA sequencing, the reads are often aligned to common exons, making it difficult to determine their origin. Although junction reads that span exons can improve the accuracy of analysis, their effectiveness is limited when transcriptional isoforms share splicing sites. ONT-IrRNA-seq (Oxford Nanopore Technologies long-read RNA sequencing) can directly detect full-length isoforms, capture unique transcripts, eliminate or reduce detection biases, and improve the accuracy of the analysis of differentially expressed transcripts.
advantages
✔ Improve gene annotation: Discover new transcripts and genes and obtain a comprehensive transcriptome map.
✔ Differential analysis: Differentially expressed genes (DEG), differentially expressed transcripts (DET), differentially used transcripts (DTU).
✔ Gene structure analysis: Accurately analyze alternative splicing (AS), polyadenylation sites (APA), and the length of 3’UTR.
✔ Transcript resolution: Analyze the regulatory mechanisms and functions of gene expression at the transcript level.
workflow
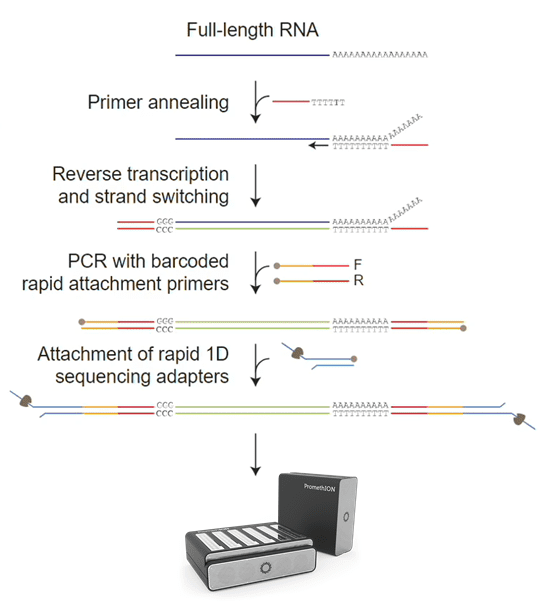
Firstly, RNA containing a polyA tail is captured by olig dT magnetic beads. Then, the first strand of cDNA is synthesized by reverse transcription using a primer composed of olig dT plus a fixed sequence. Next, the TSO sequence is added to the 3’ end of the cDNA through strand displacement. After that, primer PCR amplification is carried out to form double-stranded cDNA. Finally, the ends of the double-stranded cDNA are blunted and ligated with ONT sequencing adapters to form a sequencing library.
research Cases
case 1
Title: Long-read sequencing reveals the landscape of aberrant alternative splicing and novel therapeutic target in colorectal cancer
Journal: Genome Medicine
Impact Factor (IF): 10.4
Samples: 76 colorectal cancer tissues and 10 normal tissues
Technical Method: Oxford Nanopore Technologies (ONT) long-read sequencing
Alternative splicing plays a crucial role in carcinogenesis and cancer progression. By combining long-read sequencing with short-read RNA-seq to study the complexity of the colorectal cancer (CRC) transcriptome, 90,703 transcripts were discovered, and more than 62% of them were novel. The new transcripts were mostly sample-specific, had low expression levels, contained multiple exons, and showed obvious oncogene characteristics. 1,472 differentially expressed alternative splicing events were associated with the prognosis of CRC, and the new subtypes might affect patient survival. TIMP1 Δ4-5 was downregulated in CRC and could inhibit tumor growth and metastasis. SRSF1 maintained the inclusion of exons 4-5 of TIMP1. The CRISPR/dCasRx strategy could suppress the growth of CRC. The new findings provide resources for CRC research, and TIMP1 Δ4-5 may be a potential therapeutic target
case 2
Title: Full-Length Immune Repertoire Reconstruction and Profiling at the Transcriptome Level Using Long-Read Sequencing
Journal: Clinical Chemistry
Impact Factor (IF): 7.1
Samples: Peripheral blood from 8 patients with acute lymphoblastic leukemia, 3 patients with allergic diseases, 4 patients with psoriasis, and 5 patients with prostate cancer.
Technical Method: Oxford Nanopore Technologies (ONT) long-read sequencing
The diversity of the immune repertoire makes it challenging to reconstruct the full-length immune repertoire using traditional sequencing. The authors developed a full-length immune repertoire sequencing (FLIRseq) workflow based on linear rolling circle amplification and nanopore sequencing. FLIRseq is superior to BCR/TCR-seq, as it can provide long CDR3, B cell isotype and rarely used V gene sequences. FLIRseq has observed an increase in the diversity of clonotypes and a decrease in the percentage of abnormal BCR/TCR in patients during the remission period of leukemia. For patients with allergic diseases or psoriasis, FLIRseq provides direct insights into V(D)J recombination and specific immunoglobulin classes. FLIRseq is capable of conducting unbiased and comprehensive analyses of direct V(D)J recombination and immunoglobulin classes, which helps to characterize pathogenic mechanisms, monitor minimal residual disease and customize adoptive cell therapies.