

RXBio Translates Sequence to Science and Industry

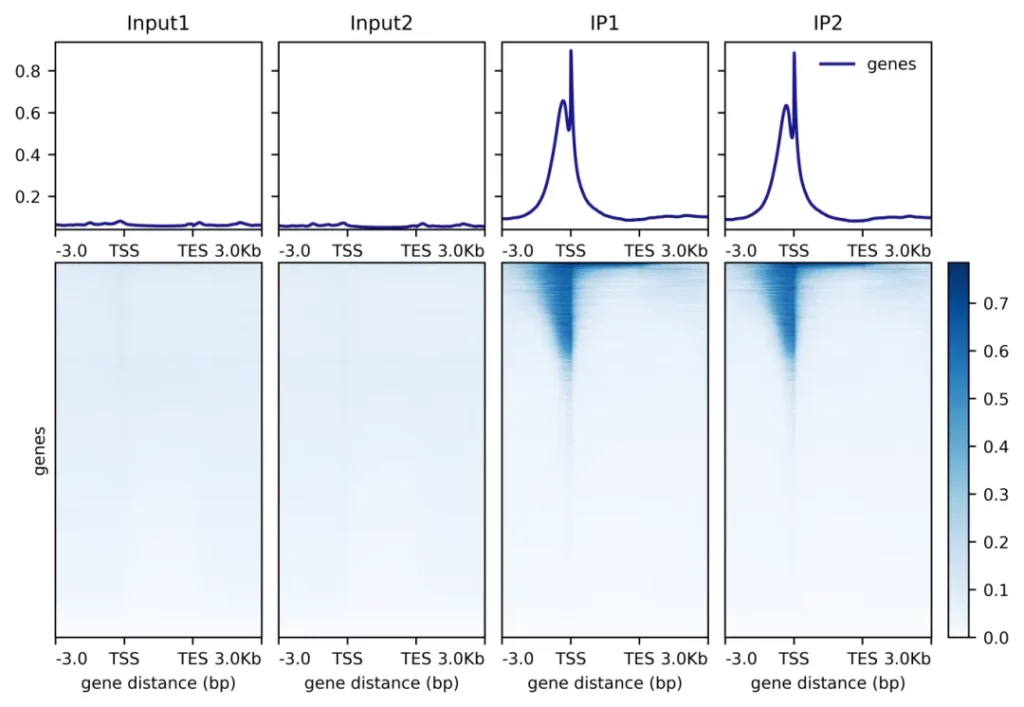
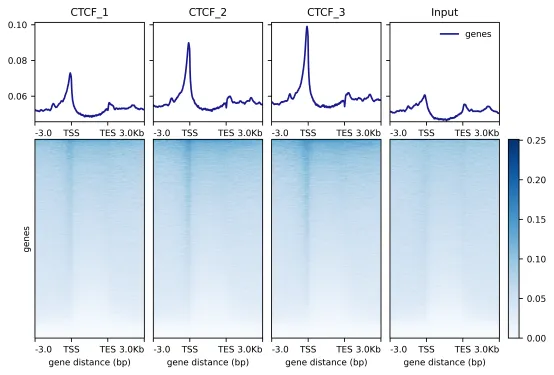
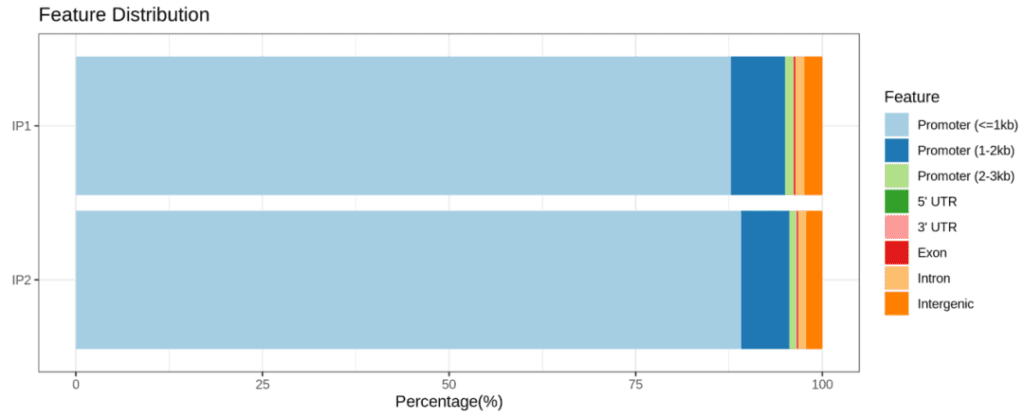
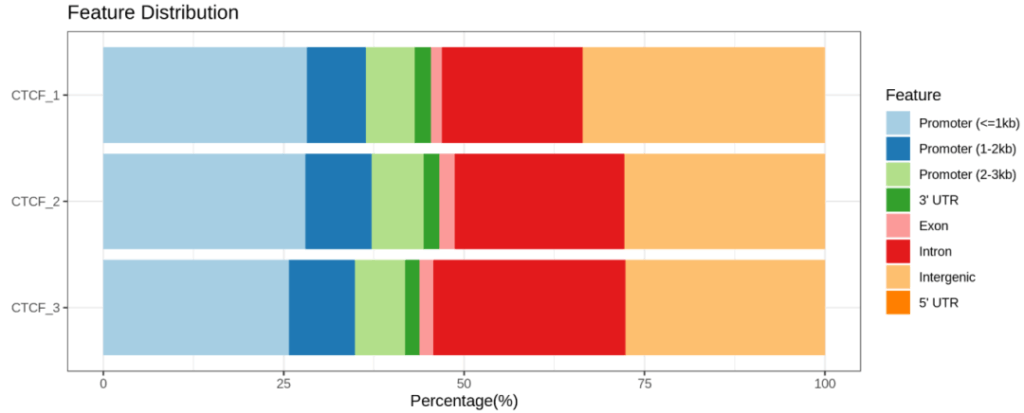
The gene expression of eukaryotes is regulated by RNA polymerase, transcription factors, histone modifications, and so on. Epigenetic researchers have proposed the chromatin immunoprecipitation technique (ChIP) to locate the regions of DNA-protein interactions. With the development of next-generation sequencing technology, the combination of ChIP and sequencing has led to the development of the ChIP-seq technique. Matching the binding footprints of transcription factors at single-base resolution is crucial for studying the regulatory mechanisms of transcription factor binding. However, the traditional ChIP-seq technique has limited resolution and cannot determine specific binding sites. To determine specific binding sites, it is necessary to predict the binding sites and conduct single-base mutations, and then construct a large number of reporter gene vectors for verification. These methods involve a large amount of work, a long cycle, and a high false positive rate.
The team led by Professor Frank Pugh at Pennsylvania State University Park has developed the ChIP-exo technique to locate the binding sites of transcription factors across the whole genome at single-nucleotide resolution. Compared with ChIP-seq, ChIP-exo requires steps such as DNA end repair, addition of an adenine (A), and ligation of adapters, which reduces the amount of recovered DNA. DNA fragments need to undergo two inefficient ligations to obtain adapters at both ends. Low starting amounts of ChIP DNA are prone to over-amplification during the PCR process for library preparation, resulting in non-reproducible noisy data.
After independent development and optimization by Wuhan Ruixing, it takes advantage of the fact that digestion of DNA by lambda exonuclease will generate single-stranded DNA (ssDNA). By constructing a library using single-stranded DNA as a template, the reaction steps such as DNA end repair, addition of an A, ligation of adapters, and rinsing operations that are required before digestion by lambda exonuclease can be omitted. This increases the amount of recoverable DNA and improves the success rate and yield of library construction.
✔ High resolution: Achieving single-base resolution.
✔ High specificity: Exonuclease digestion reduces background DNA.
✔ High signal-to-noise ratio: Eliminating a large amount of noise generated by the detection system.
✔ Simple process: Optimizing the library construction steps to improve the success rate.
1) Formaldehyde cross-linking;
2) Sonication fragmentation;
3) Immunoprecipitation;
4) Exonuclease digestion;
5) DNA extraction;
6) Library construction using single-stranded DNA.
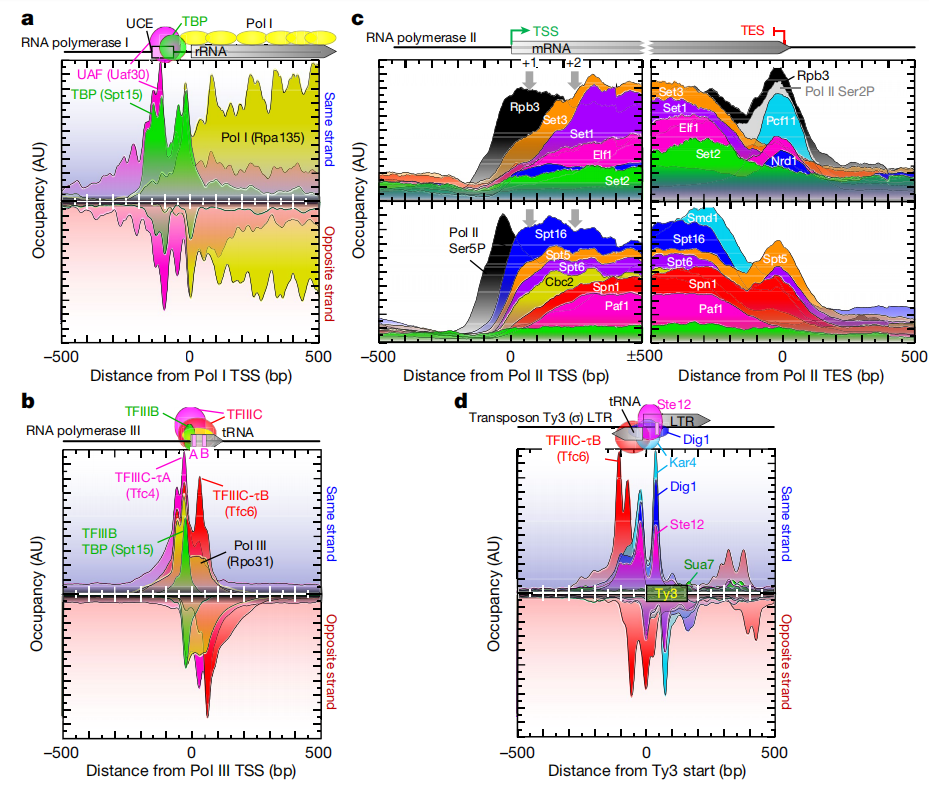
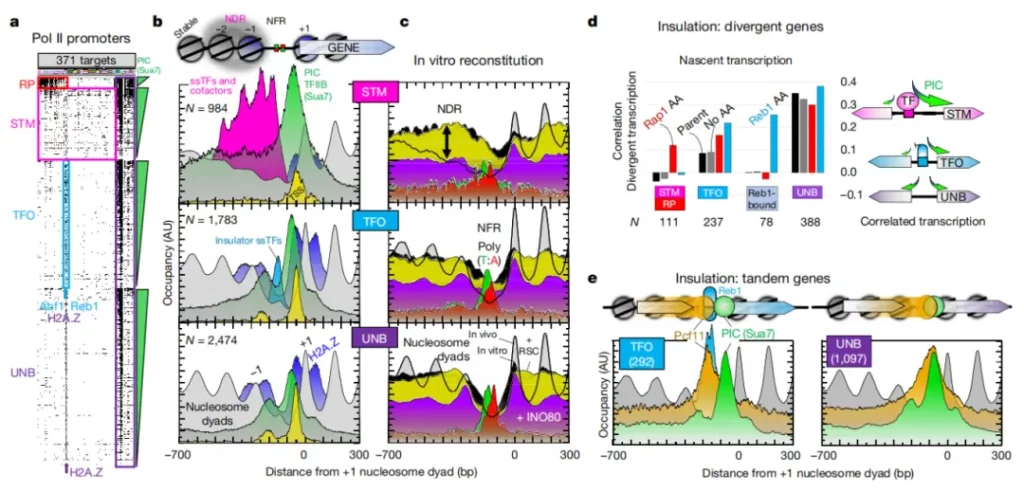
The latest research in an article titled “A high-resolution protein architecture of the budding yeast genome” published in the journal Nature has discovered the precise binding sites of more than 400 different chromosomal proteins in the yeast genome, and most of them can regulate gene expression.
The team completed over 1,200 ChIP-exo experiments and generated billions of data points. To analyze these data, they utilized the supercomputing cluster at Pennsylvania State University and their self-developed bioinformatics tools to identify and reveal the organizational patterns of regulatory proteins in the yeast genome. The study found that a few unique combinations of proteins in the yeast genome are reused.
The researchers discovered that RNA acts contrary to the activity of TOP1. Importantly, RNA polymerase II (a multi-protein complex that can transcribe DNA into mRNA) can activate the transcription process. Researcher Mannan Bhola said that this study has revealed a unique mechanism in which RNA can regulate DNA relaxation mediated by TOP1, thus playing an important role in regulating the gene transcription process. By identifying that TOP1 can act as an RNA-binding protein, the researchers have provided new insights into the interaction between RNA and DNA during transcription. TOP1 is overexpressed in many human cancers and can thus help regulate the function of DNA during the transcription process, providing certain information for the development of new cancer therapies.
The authors used the ChIP-exo/seq technique to define specific structures in Saccharomyces cerevisiae. We identified 21 meta-combinations containing approximately 400 different proteins, which are related to DNA replication, centromeres, sub-centromeres, transposons, and the transcription of RNA polymerase (Pol) I, II, and III. Replication proteins engulf nucleosomes, there are no nucleosomes at the centromere, and inhibitory proteins have three nucleosomes at the subtelomeric X element. Most of the Pol II-related promoters lack regulatory regions and only contain core promoters. These promoters contain a short nucleosome-free region (NFR) adjacent to the +1 nucleosome and bind to the transcription initiation factor TFIID to form a pre-initiation complex. This study has discovered two different gene regulatory structures, expanding the traditional model. Constitutive genes perform basic functions and require simple regulation; inducible genes are sensitive to environmental signals and have a more complex structure. The traditional model involves transcription factors binding to DNA to control expression. However, most “housekeeping” genes in yeast lack the specific protein-DNA structures that are characteristic of inducible genes.
Reference
[1]Rossi, M.J., Lai, W.K.M. & Pugh, B.F. Simplified ChIP-exo assays. Nat Commun 9, 2842 (2018). https://doi.org/10.1038/s41467-018-05265-7.
[2]Rossi, M.J., Kuntala, P.K., Lai, W.K.M. et al. A high-resolution protein architecture of the budding yeast genome. Nature 592, 309–314 (2021).https://doi.org/10.1038/s41586-021-03314-8.