
introduction
RIP-seq studies the interaction state between the most primitive RNA-binding proteins (RBPs) and RNA under natural conditions. The captured RNA is not fragmented by enzymatic digestion, so the obtained RNA fragments are relatively long. Moreover, there is no need for the gel cutting and purification steps for RBPs. Its operation process is simple and has a high success rate, but it cannot obtain the RNA sites directly bound by RBPs.
iRIP-seq (Improved RIP-seq) is a new technology independently developed by our company for researching RBPs. It incorporates the UV-crosslinking and MNase digestion experiments in the CLIP-seq technology into the traditional RIP technology. Meanwhile, it uses the data analysis process of CLIP-seq to obtain the peaks and motif information of RBP binding. This technology can accurately identify the RNA binding sites while retaining the simplicity of RIP-seq.
advantages
The advantages of the iRIP-seq technology lie in its high efficiency and specificity, enabling precise analysis of RNA-binding proteins (RBPs) in the target biological systems under study. This technology can not only successfully detect the known interactions between RNA and proteins but also discover new RNA-binding proteins and the interaction networks they are involved in. In addition, this technology helps us reveal the importance of RNA modifications in the processes of transcriptional and post-transcriptional regulation, deepening our understanding of the interactions between RNA and proteins. Overall, iRIP-seq provides strong support for the study of the interactions between RNA and proteins, allowing us to gain a deeper understanding of the complex interaction networks between RNA and proteins in biological systems. This understanding not only helps to uncover the mysteries of gene expression regulation but also offers new perspectives and directions for the analysis of disease mechanisms and the development of drugs.
Principle
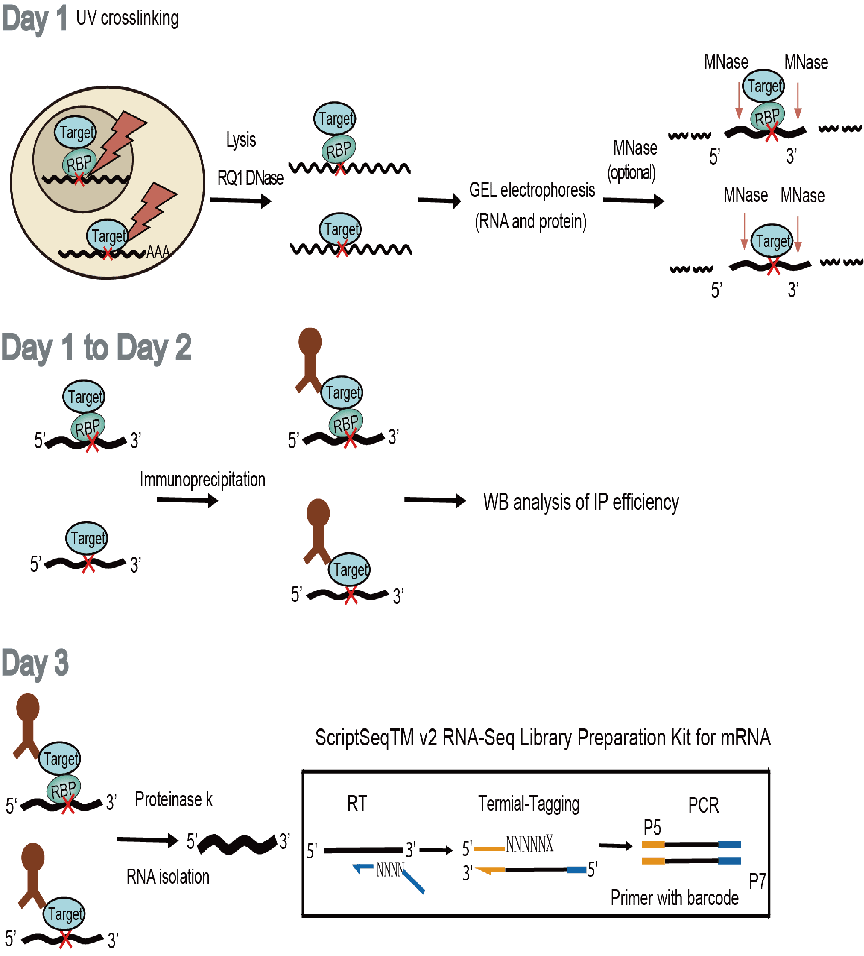
The core steps of this technology include an added UV-crosslinking step. This step fixes the interaction sites between RNA-binding proteins (RBPs) and RNA by creating covalent bonds, enabling the interaction sites to be identified in a more stable and precise manner. During the process of enzymatically fragmenting RNA, the micrococcal nuclease (MNase) is employed. This enzyme has the characteristic of being calcium ion-dependent and its enzymatic reaction can be terminated by chelating agents such as EDTA. In addition, stricter ionic detergent and high-salt rinsing conditions result in less noise during the experiment and improve the signal-to-noise ratio of the experimental results. Finally, the improved library construction method is also one of the highlights of this technology. By successfully constructing a library of reverse transcription (RT) products with interrupted UV-crosslinking sites, the detection of the interaction sites between RBPs and RNA becomes more precise and efficient.
Research Cases
▶ The FOXN3-NEAT1-SIN3A repressive complex promotes the progression of hormone-responsive breast cancer.

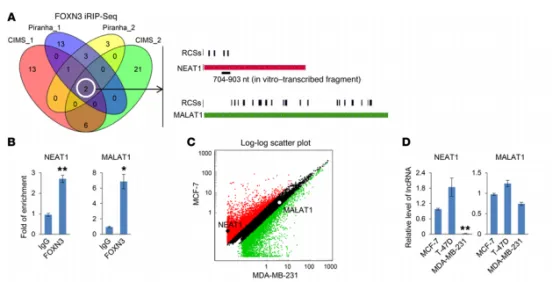
The team led by Academician Shang Yongfeng published a paper titled “The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer” in JCI (with an Impact Factor of 15.9), reporting the role of the transcription factor FOXN3 in breast cancer metastasis and its underlying mechanism.
In MCF-7 cells, the method of improved RNA immunoprecipitation coupled with high-throughput sequencing (iRIP-seq) was used to identify the RNAs that could bind to FOXN3. It was verified that the interaction between FOXN3 and the SIN3A complex requires the involvement of a long non-coding RNA (lncRNA) named NEAT1, which is induced by estrogen.
The combined analysis of the results from chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) and the data from capture hybridization analysis of RNA targets by deep sequencing (CHART-seq) demonstrated that the FOXN3/NEAT1/SIN3A complex transcriptionally represses a series of important genes involved in breast cancer metastasis, including GATA3 and TJP1.
▶ MEX3B inhibits the production of collagen in eosinophilic nasal polyps by downregulating the stability of TGFBR3 mRNA in epithelial cells.

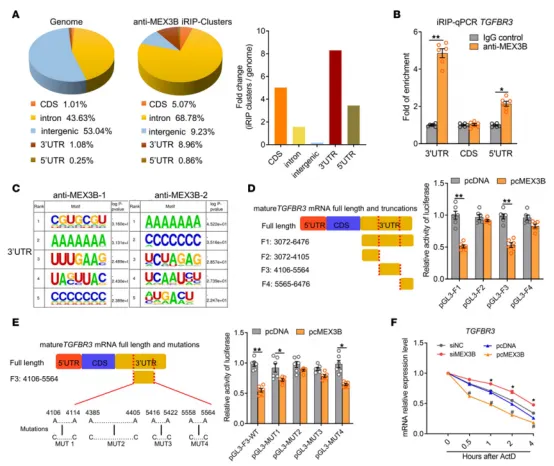
The team led by Professor Liu Zheng, the vice president of Tongji Hospital affiliated with Tongji Medical College of Huazhong University of Science and Technology, published a research titled “MEX3B inhibits collagen production in eosinophilic nasal polyps by downregulating epithelial cell TGFBR3 mRNA stability” on the cover of JCI insight (with an Impact Factor of 8.0).
To explore the direct downstream targets of MEX3B as an RNA-binding protein (RBP) in airway epithelial cells, the authors conducted RNA-Seq and iRIP-Seq after knocking down and overexpressing MEX3B. A total of 7 overlapping genes were obtained, and the overlapping mRNAs were considered to be the mRNAs directly bound and regulated by MEX3B. Among them, TGFBR3 attracted particular attention. TGF-βR3 can bind with high affinity and enhance the function of the TGF-β2 pathway. In non-eosinophilic nasal polyps, the expression of TGF-βR3 is decreased, resulting in the weakened function of the TGF-β2 pathway, the reduced expression of genes related to the collagen family, and the promotion of the formation of tissue edema.
▶ IGF2BP2 regulates macrophage phenotypic activation and inflammatory diseases by stabilizing TSC1 and PPARγ.


The research team led by Professor Li Jingxin from the School of Basic Medical Sciences at Shandong University published a research paper titled “The m6A Reader IGF2BP2 Regulates Macrophage Phenotypic Activation and Inflammatory Diseases by Stabilizing TSC1 and PPARγ” in Advanced Science (with an Impact Factor of 15.1).
By utilizing the iRIP-seq technology, it was found that in macrophages, IGF2BP2 can bind to the mRNAs of multiple genes that play important roles in the process of macrophage activation and polarization, such as Kcnn4, Arg1, Hist1h1b, Hist1h4c, Cd74, Ak2, H2-Eb1, Csf1r, and PPARγ, and participate in the regulation of the expression of TSC1 and PPARγ through m6A modification. IGF2BP2 tilts macrophages from a pro-inflammatory phenotype to an anti-inflammatory phenotype through the TSC1-mTORC1 pathway and the fatty acid metabolism mediated by PPARγ.
▶ The iRIP-seq technology explored the transcriptome-wide binding profile of IGF2BP2 in Jurkat cells.

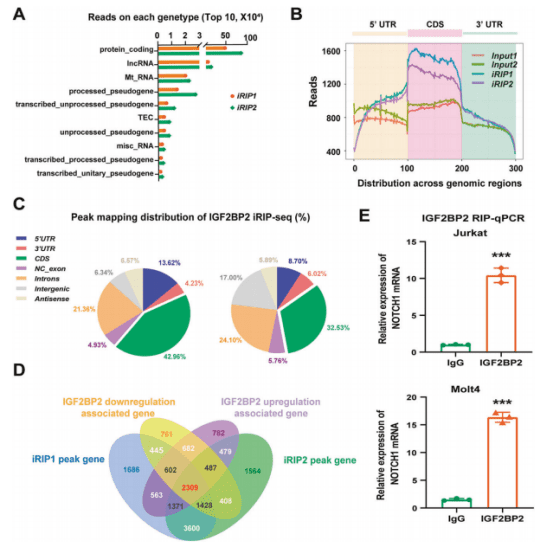
The research team led by Professor Li Jingxin from the School of Basic Medical Sciences at Shandong University published another article titled “Inhibition of the m6A reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia” in the journal Leukemia (with an Impact Factor of 11.4). They utilized the iRIP-seq technology to explore the transcriptome-wide binding profile of IGF2BP2 in Jurkat cells. Most of the reads bound by IGF2BP2 were located in mRNA and were highly enriched in the coding sequence (CDS).
Then, RNA-seq sequencing was performed on Jurkat cells with IGF2BP2 upregulated or downregulated. Combining with the iRIP-seq data, 2309 regulatory genes were identified, and a large number of genes (NOTCH1, NKX2-1, BCL11B, RUNX1, FBXWT, STAT5B, DNM2, AKT1/2, MTOR, ABL1, DNMT3A and RPL11) were related to the pathogenesis of T-cell acute lymphoblastic leukemia (T-ALL). As an m6A reader, IGF2BP2 increased the stability of NOTCH1 during the methylation process, maintained and increased the growth of transformed T-ALL cells. The inhibitor JX5 targeting IGF2BP2 activity effectively inhibited the activation of NOTCH1 and the growth of T-ALL.
▶ A-to-I RNA editing in bacteria increases pathogenicity and tolerance to oxidative stress.

The joint team of Zhu Bo and Chen Gongyou from the School of Agriculture and Biology at Shanghai Jiao Tong University published a research finding titled “A-to-I RNA editing in bacteria increases pathogenicity and tolerance against oxidative stress” in PLoS Pathogens (with an Impact Factor of 6.7), revealing a new mechanism by which prokaryotic bacteria adapt to environmental changes through A-to-I RNA editing.
The research team used iRIP-seq data and a self-developed Python script to identify and analyze the A-to-I editing events in Xanthomonas oryzae pv. oryzicola (Xoc) under oxidative stress. It was found that under the stress of oxidative stress, the A-to-I editing changed the 128th amino acid of the flagellin FliC from serine (S) to proline (P), resulting in a change in the structure of the flagellar filament. This post-transcriptional editing can increase the tolerance to oxidative stress and the virulence on rice by increasing the formation of biofilms.