RXBio Translates Sequence to Science and Industry

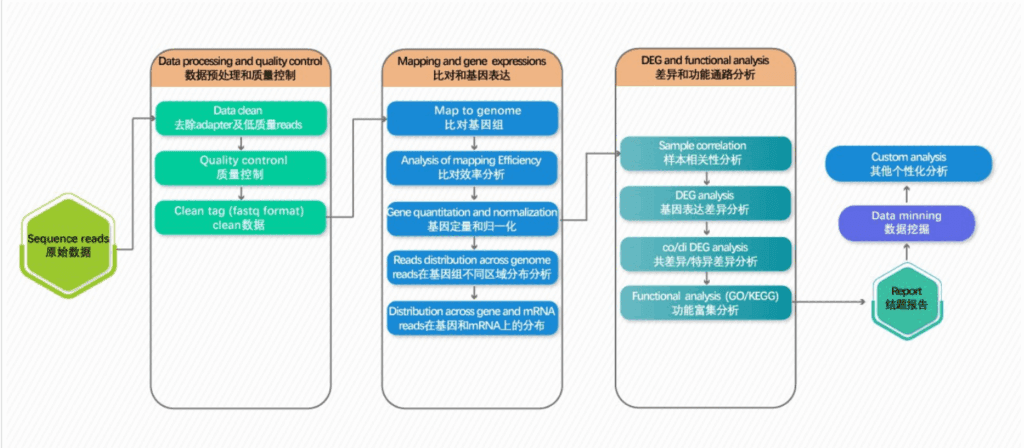
Transcriptome sequencing (mRNA-seq)
Messenger RNA (mRNA) is a type of single-stranded RNA that is transcribed from one strand of DNA as a template, carries genetic information, and can direct protein synthesis.
mRNA sequencing uses the oligod(T) enrichment library construction method combined with high-throughput sequencing to study the expression and regulation of mRNA, and is widely applied in various basic research.
Whole transcriptome sequencing
Unlike transcriptome sequencing that can only obtain mRNA information, whole transcriptome sequencing can simultaneously sequence and analyze pre-mRNA, lncRNA, circRNA or microRNA separately through two different library construction methods. It can also conduct combined analysis of multiple types of RNA, integrated analysis of endogenous competing RNA, screening of molecular markers, etc., to explore their potential regulatory network mechanisms and comprehensively reveal the issues of transcriptional regulation.
Ribosomal RNA (rRNA) is the type of RNA that is present in the largest amount in cells. However, during sequencing, a large quantity of rRNA can overshadow the sequencing abundance of other genes. Therefore, it is necessary to remove rRNA to construct strand-specific libraries in order to obtain data on lncRNA, mRNA and circRNA.
Compared with the other three types of RNA (pre-mRNA, lncRNA, circRNA), miRNA has a relatively short sequence, only 18 to 26 nucleotides. Therefore, the method for constructing libraries is different, and it is necessary to conduct sequencing on the sequencing instrument separately.
✔ Multiple imported/domestic sequencing platforms
✔ In-house analysis processes
✔ Advanced data analysis
✔ Support for personalized services
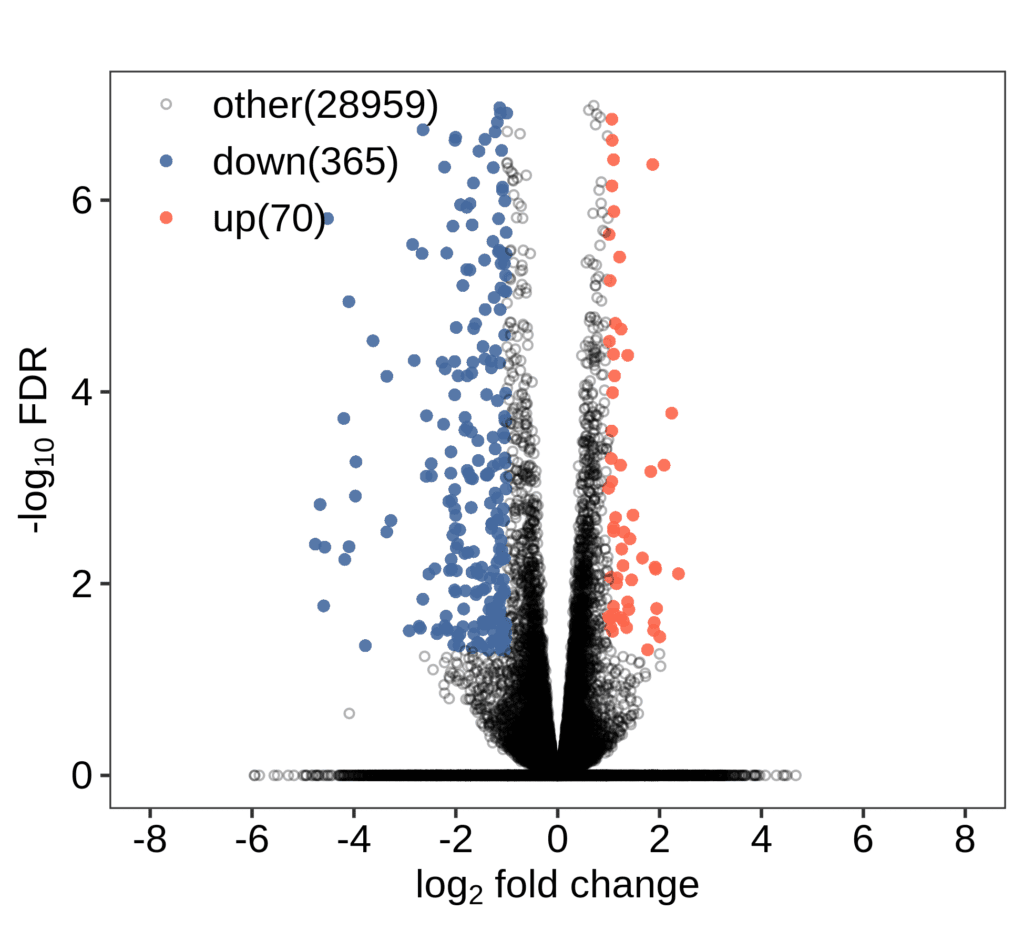
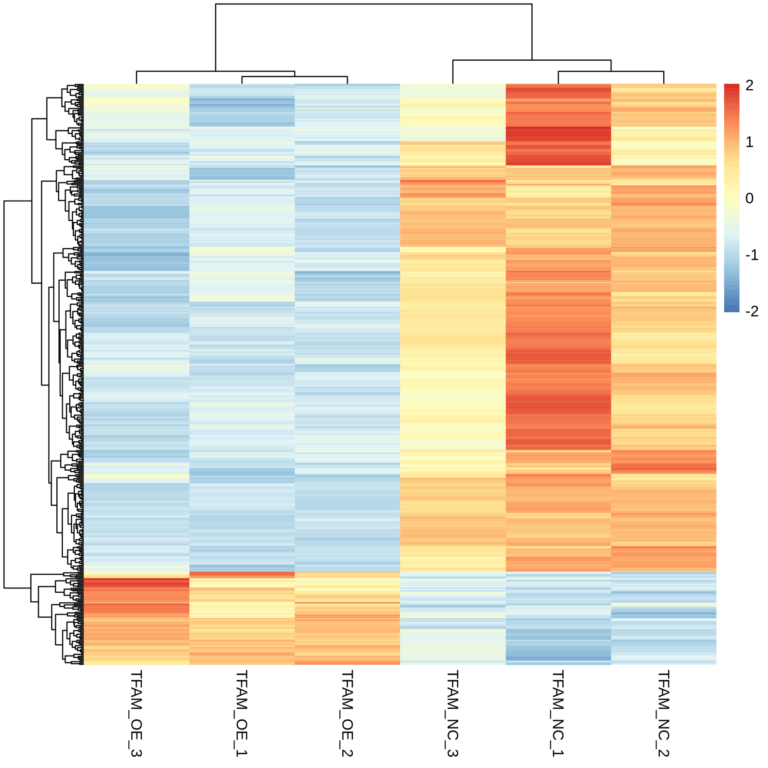
Transcriptome analysis revealed distinct gene expression patterns between the healthy group and the chronic myeloid leukemia (CML) patient group.
Journal: Journal of Advanced Research
Impact Factor: 12.822
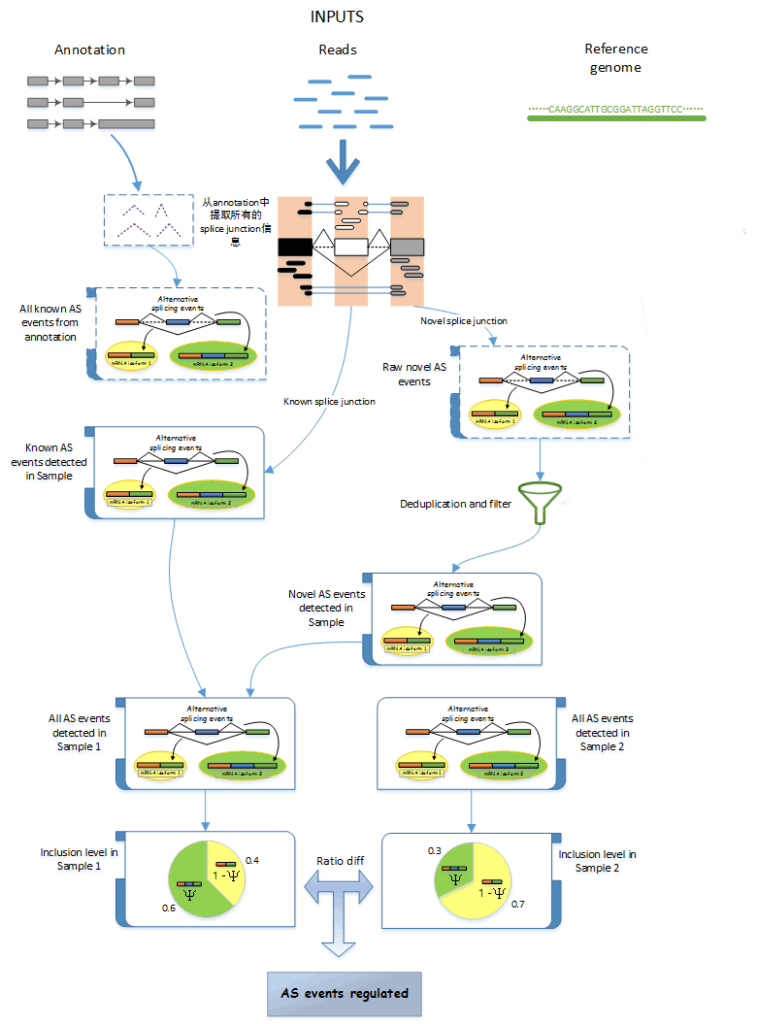
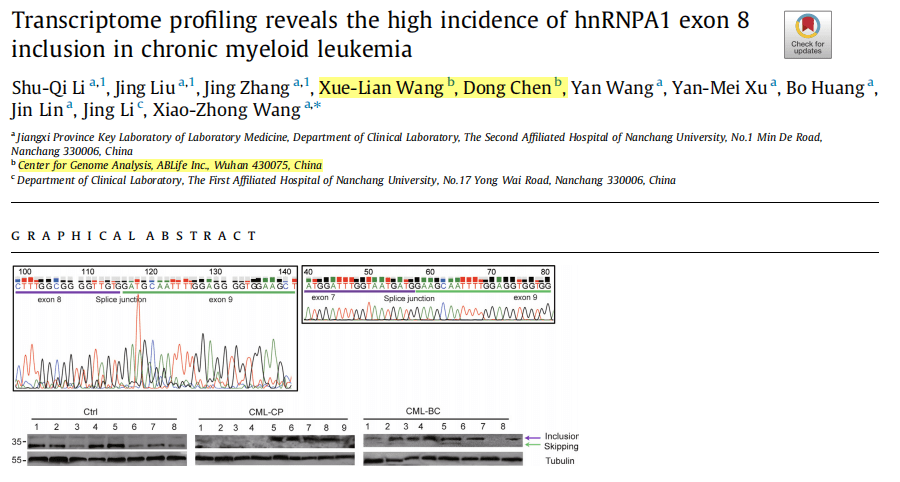
This study utilized transcriptome sequencing technology to reveal the transcriptome profiles of patients with chronic myeloid leukemia at different stages. The ABLas alternative splicing analysis process was employed to compare the alternative splicing regulatory patterns in two types of patients. Patients with leukemia in the blast crisis phase exhibited more significant splicing dysregulation. The auxiliary splicing factor hnRNPA1 was screened out and could serve as a biomarker to distinguish between leukemia patients and healthy individuals. Experiments demonstrated that the full-length splicing isoform A1-b of hnRNPA1 was significantly upregulated in patients in the blast crisis phase. This unique expression pattern might provide a new prognostic indicator for the classification of acute and chronic leukemia patients.